Fluorine Notes, 2011, 77, 1-2
FOUNDATIONS AND DEVELOPMENT OF CHEMISTRY OF POLYFLUOROARENES AND -HETARENES
(dedicated To the 50th Jubilee of Laboratory of Halogen Compounds at NIOC SB RAS)
V.E. Platonov, T.D. Petrova
N.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, Siberian Branch of the Russian Academy of Sciences (NIOC SB RAS), Novosibirsk, Russian Federation, E-mail: petrova@nioch.nsc.ru
Abstract: This review summarizes the results on the chemistry of polyfluoroarenes and -hetarenes obtained in the Laboratory of Halogen Compounds of NIOC SB RAS within 50 years of its activities. These results include (i) the development of methods for synthesizing polyfluoroarenes and –hetarenes that enabled obtaining polyfluoroarenes and polyfluorochloroarenes by nucleophilic substitution of chlorine by fluorine in perchloroarenes by the action of alkali metal fluorides in the absence of solvents, (ii) the syntheses of not fully fluorinated arenes from perfluoro- and polyfluorochloroarenes by hydrogenolysis of C–F and C–Cl bonds with Zn(Cu) in aqueous DMF, as well as (iii) the methods for functionalization of perfluoroarenes via reactions with dihalogencarbene sources, or by transformations of the functional groups already contained in perfluoroarenes, by the action of nucleophilic, electrophilic, and radical reagents.
Bibliography – 95 references.
Content
I Introduction
IIThermolytic transformations
III
Polyfluoroarenes and Zn: hydrogenolysis reactions of perfluoro- and
chloropolyfluoroarenes by the action of Zn(Cu)-DMF-H2O; preparation of
hydropolyfluoroarenes. Polyfluoroaromatic organozinc compounds
IVChemistry of functional derivatives of polyfluoroarenes
V Conclusion
I. Introduction
The chemistry of polyfluoroarenes and – hetarenes has been the major scientific direction in the work
of the Laboratory of Halogen Compounds (hereinafter referred to as Laboratory) since its foundation.
The interest in compounds of this type was encouraged by the achievements in studying their chemistry
and the discovered unique properties of other polyfluoroorganic compounds. This stimulated the development
of methods for synthesizing polyfluoroarenes and –hetarenes and studies of their reactivities, which
were scarcely known until the sixties of the last century, but nevertheless such compounds also could
have been very promising. A convenient method for the synthesis of the first representative of polyfluoroarenes
was developed in the Laboratory and published in 1963 [1]. This method stimulated rapid development
of the chemistry of polyfluoroarenes and –hetarenes both in our country and abroad, until now NIOC
SB RAS is one of the leading research centres in this field. Usually the development of the synthesis
methods and studies of chemistry of polyfluoroaromatic compounds are closely interlaced in the Laboratory,
however, the initial efforts were directed specifically to the development of a convenient and efficient
method for the synthesis of the key polyfluoroaromatic compounds to make them available and get an
opportunity to study their reactivities and applications. Studying reactions of these compounds under
various conditions (thermolysis, photolysis) with reagents of various nature (nucleophiles, electrophiles,
radicals, carbenes) inevitably led to discovering new approaches to the synthesis of polyfluoroarenes
and –hetarenes, not only accumulation of knowledge about reactivity. A large team of scientists working
in the Laboratory have been carrying out these investigations over 50 years of its existence, and
the main part of the results is reflected in this review and other surveys that will be published
after this one.
This review is concerned with the methods for synthesizing polyfluoroarenes
and –hetarenes developed by the Laboratory which can be divided in three groups. The first group
includes the methods for preparation of perfluoroarenes and polyfluorochloroarenes by nucleophilic
substitution of chlorine by fluorine in perchloroarenes by the action of alkali metal fluorides without
solvent. The second group of methods that makes it possible to obtain non-fully fluorinated arenes
from perfluoro- and chloroperfluoroarenes is based on hydrogenolysis of them, i.e. substitution of
fluorines and chlorines by hydrogens by the action of Zn (Cu) in aqueous DMF. Finally, the third
group includes methods of functionalization of perfluoroarenes by the action of dihalogencarbenes,
as well as via transformations of the functional groups already contained in polyfluoroarenes
using nucleophilic, electrophilic and radical agents; this enables obtaining a wide variety of compounds
including heterocyclic derivatives. The above mentioned transformations included in this group are
so numerous that this review considers only a part of the investigations carried out by the Laboratory
in this direction.
Thermolytic transformations
1. Nucleophilic reactions. Introduction of fluorine in aromatic ring by nucleophilic substitution of chlorine for fluorine in perchloroarenes by the action of alkali metal fluorides in the absence solvents. Synthesis of perfluoro- and polyfluoroarenes
The Laboratory was the first research centre in the world where the most convenient and efficient method for the preparation of polyfluorinated aromatic compounds has been developed that remains topical today. This method is based on nucleophilic substitution of the chlorine atoms in perchlorinated arenes for fluorine under the action of potassium or cesium fluorides in the absence of solvents and at high temperature [1]. The creation of this method is based on the earlier works by some of its authors concerning exchange of chlorine atoms in chloronitrobenzenes for fluorine by reacting with alkali metal fluorides in the absence of solvents [2,3]. Activation of the chlorine with the nitro group and the elevated temperature favoured these reactions. The reaction of hexachlorobenzene with potassium fluoride in an autoclave at 460 – 470 °C gave the complete exchange product – hexafluorobenzene – in 20 – 22% yield and a great amount of the products of incomplete exchange including chloropentafluorobenzene which is the most important among them (24%) [1]. When the temperature was elevated to 530 °C, a mixture of hexafluorobenzene and chloropolyfluorobenzenes was obtained in yield up to 70% and the content of hexafluorobenzene increased to 45%. The mixture was divided in components by rectification. Hexafluorobenzene, chloropentafluorobenzene and fractions of compounds with the lesser degree of replacement of chlorine by fluorine were isolated in yields that made these compounds available both as subjects of research and for industrial applications [1,4]. Solid 1,3,5-trichlorotrifluorobenzene was isolated from the trichlorotrifluorobenzene fraction in 60% yield.
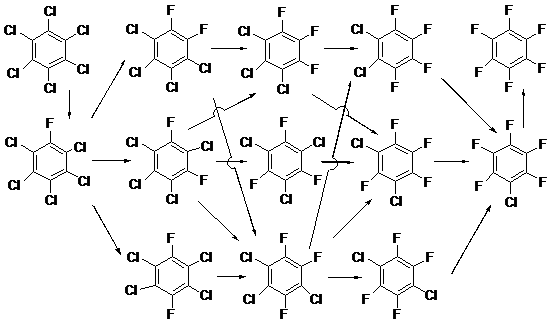
Exchange of chlorine atoms for fluorine in perchloroarenes under the action of potassium fluoride in the absence of solvent proved to be a general method for the preparation of perfluoroaromatic analogues. Octafluoronaphthalene [5], decafluorodiphenyl [5], and decafluoropyrene [6] have been synthesized by such a way.
This method is also suitable for obtaining some functional derivatives of polyfluoroarenes, in particular, polyfluoroaromatic acid fluorides. It is reasonable that in this case, the exchange reaction occurs under the milder conditions as a result of the additional activating effect of electron-acceptor carbonyl moieties [7-9]. Exchange of chlorine for fluorine have been also carried out in polychlorinated quinones. 1,2,3,4-Tetrafluoroanthraquinone has been synthesized from the tetrachloroderivative by this method in 92% yield [10]. The method proved to be suitable even for incompletely chlorinated non-activated arenes including chlorobenzene, but in this case, the essential factor is a decrease in thermal stability of both starting compounds and final products, containing hydrogen atoms that stipulates a decrease in yields of the exchange products as well as occurrence of the more complicated transformations to form isomers and products of reductive dehalogenation [11].
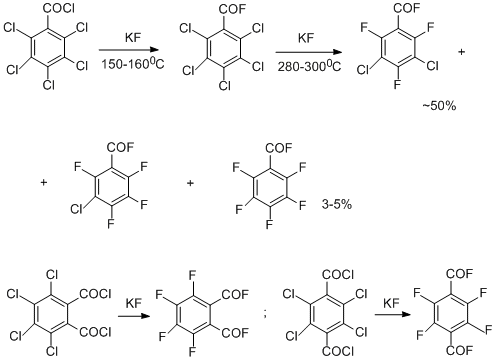
Since its creation, the thermal method for the synthesizing polyfluoroarenes by replacement of chlorine for fluorine is widely used worldwide (Halex process) for obtaining compounds of diverse types including a wide variety of polyfluorinated heterocyclic systems [12]. Up to now this method remains the best one for the synthesis of hexafluorobenzene and is used for its commercial production, although the method has been modified and improved by the efforts of numerous research centres and companies; at present such efforts are continued and directed to searching for effective phase transfer catalysts, preparing the potassium fluoride having highly developed surface, and decreasing the reaction temperature, etc.[13].
It should be noted that the reaction mixture obtained in the reaction of hexachlorobenzene with potassium fluoride at high temperatures and in the absence of solvent, contained small amounts of perfluorinated benzene homologues such as toluene, isomeric xylenes, mesitylene, as well as perfluoroindan. The formation of these compounds was assumed to be associated with involving difluorocarbene in the process [14,15], this assumption stimulated a series of studies on reactions of perfluoroarenes and their derivatives with dihalogencarbenes.
2 Reactions with dihalocarbene sources
2.1 Reactions with difluorocarbene sources. Trifluoromethylation, cycloalkenylation, polyfluorovinylation of polyfluoroarenes, synthesis of polyfluorinated heterocyclic compounds
To determine the most probable routes for the formation of perfluorinated trifluoromethyl derivatives in the reaction of hexachlorobenzene with KF (see above), hexafluorobenzene was reacted with КF under similar conditions [16]; it was shown that the reaction products were the same trifluoromethyl derivatives as in the case of hexachlorobenzene. At the first step hexafluorobenzene appears to react with KF to form a σ-complex, which decomposes to generate difluorocarbene. The latter, in its turn, reacts with the starting hexafluorobenzene to give octafluorotoluene and then other trifluoromethylation products according to the similar scheme [16].
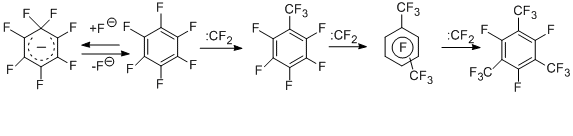
The carbene scheme for the formation of trifluoromethyl derivatives is also confirmed by the results obtained in the reactions of perfluoroarenes with the notorious difluorocarbene sources, namely, polytetrafluoroethylene (teflon) and tetrafluoroethylene. The data for hexafluorobenzene are presented in the scheme below [17,18].
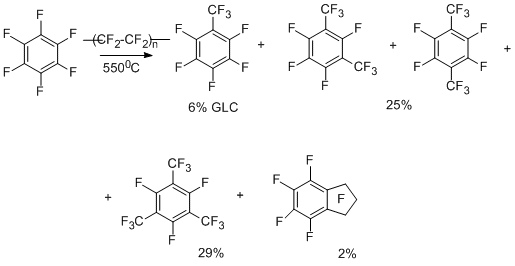
When the reaction with tetrafluoroethylene is carried out at the higher temperature (720–770°C) [19], the conversion degree and the yields of products increase (the yield of octafluorotoluene is 65 to 67%). Pentafluoropyridine [20], decafluoroindan [21], decafluorodiphenyl [21], and octafluoronaphthalene [21] also undergo trifluoromethylation.
It is essential that in the formation of trifluoromethyl derivatives, meta-orientation is observed as a rule; this is consistent with electrophilic character of difluorocarbene in the above transformations. The predominant formation of 2-trifluoromethyl derivative in the case of octafluoronaphthalene, instead of the more expected for electrophilic reagents 1-isomer could be brought about the influence of thermodynamic factors on the trifluoromethylation and as a result the more thermodynamically stable 2-isomer is accumulated in the mixture. The steric hindrances for the attack of difluorocarbene at α-position of octafluoronaphthalene should not also be excluded [19]. Trifluoromethylation is a reversible process, and this reversibility enables to consider more reasonably the influence of thermodynamic factors on the trifluoromethylation reaction. For example, when octafluorotoluene is heated, its disproportionation occurs to form hexafluorobenzene and perfluoroxylenes (with meta-isomer predominating [22]), isomeric xylenes undergo disproportionation to give octafluorotoluene and perfluoromesitylene, and their "interconversion" is observed additionally [23].
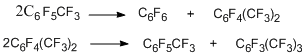
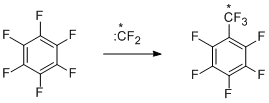
The regularities observed in the processes of disproportionation make it possible to conclude that the electrophilic difluorocarbene can be responsible for these reactions. The data obtained on trifluoromethylations, especially the results concerning the formation of octafluorotoluene from hexafluorobenzene and a labelled difluorocarbene source (fluoroform enriched by 13C isotop (46%) was used as such a source [24]) suggest that the probable route to give trifluoromethyl derivatives is insertion of difluorocarbene into C-F bond.
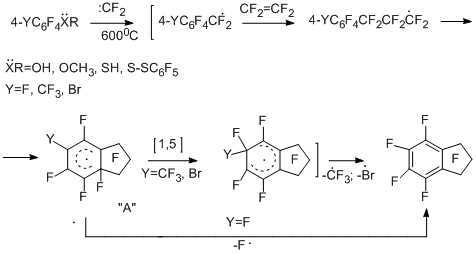
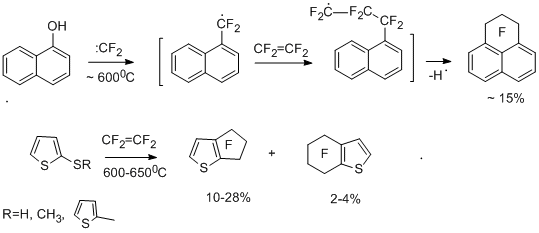
Introduction of functionalities in a perfluoroaromatic compound effects on the behaviour of polyfluoroarenes in their reactions with difluorocarbene sources. In the case of electron-donor substituents, the reactivity increases, but the reaction occurs mainly at the functional group. When electron-acceptor substituents are introduced, the reactivity decreases, and in some cases, the direction of the reaction changes. In the case of chloropentafluorobenzene and tetrafluoroethylene the products corresponding to insertion of difluorocarbene into C–Cl and C–F bonds are formed, namely, heptafluorobenzyl chloride in a mixture with chloroheptafluorotoluenes in addition to perfluoroaryltrifluoromethyl derivatives and perfluoroindan; whereas bromo- and iodopentafluorobenzenes react more unambiguously to give mainly perfluoroindan [19]. 3-Chlorotetrafluoropyridine and 5-chlorotetrafluoropyrimidine react with tetrafluoroethylene at 820°C to give trifluoromethyl derivatives of perfluoropyridine and -pyrimidine [19]. Polyfluorinated benzene derivatives, wherein the functional groups contain lone electron pairs, react with tetrafluoroethylene and other difluorocarbene sources at the lower temperature to give perfluoroindan as the major product [25,26]. It is of interest that perfluoroindan is also formed in copyrolysis of tetrafluoroethylene with perfluoro-p-cresol [26], p-bromotetrafluoroanisole [19], and perfluoro-p-thiocresol [26]. This can be explained by isomerization of radical s-complex "А" as a result of 1,5-migration of the fluorine atom and the easier elimination of radicalsCF3· and Br· from the complex because of the lower energy of C –C and C–Br bonds in comparison with C-F bond.
Probably, perfluorobenzyl radicals are involved in the formation of perfluoroindan, then they react with tetrafluoroethylene to lead to intramolecular cyclization of the radical type. Actually, under similar conditions, copyrolysis of tetrafluoroethylene with heptafluorobenzyl bromide, a notorious source of heptafluorobenzyl radical, gives perfluoroindan [26] in high yield. The assumption of the formation of heptafluorobenzyl radicals is also confirmed by the data on copyrolysis of pentafluorophenol with tetrafluoroethylene in the presence of either bromine, or 1,2-dibromotetrafluoroethane, which is capable of debrominating under the reaction conditions to form tetrafluoroethylene. As a result of these reactions, heptafluorobenzyl bromide and perfluorobenzylphenyl ether have been obtained [27].
The possible route for the formation of heptafluorobenzyl radical is presented below by the following simplified scheme in the example of the reaction of pentafluorophenol. This route is supposed to include insertion of difluorocarbene into the C-O bond [26].
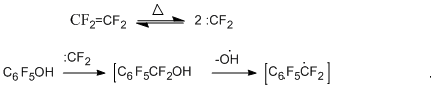
The above-mentioned polyfluorocycloalkenylation of polyfluoroarenes involving radicals of the α,α-difluorobenzyl type in the aromatic series is of sufficiently general character. Thus, copyrolysis of 4-methoxytetrafluoropyridine with tetrafluoroethylene gives perfluoro-2-pyrindan as the major product [28].
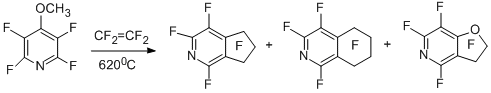
Polyfluorocycloalkenylation also takes place in reactions of non-fluorinated aromatic oxygen- and sulfur-containing derivatives [29-32].
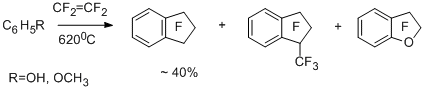
Essentially, the reactions of pentafluorotoluene and toluene with tetrafluoroethylene occur involving pentafluorobenzyl and benzyl radicals to afford 1,1-dihydrooctafluoroindan and 1,1-dihydro-2,2,3,3-tetrafluoroindan [33,34]. The more electron-donating 2-methylthiophene forms 4,4,5,5,6,6-hexafluorocyclopenta[b]thiophene as a result of involving 2-thienyldifluoromethyl radical [34,35].
The regularities observed in the reactions of polyfluoroarenes, having electron-acceptor chloroalkyl groups, with difluorocarbene sources differ from those described above. The result of these reactions is the formation of fluorovinylation products. For example, the copyrolysis of pentafluorobenzotrichloride or its para-substituted derivatives with tetrafluoroethylene gives polyfluorinated styrenes [36-38]. The process occurs via insertion of difluorocarbene into the C–Cl bond at the benzylic position followed by dechlorination or dehydrochlorination of the intermediate polyfluorochloroethyl derivatives [36-38].
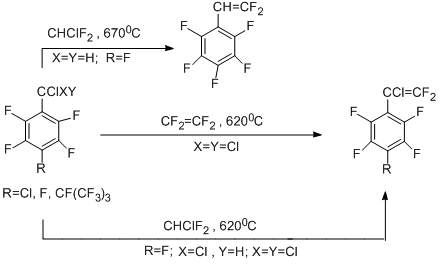
2.2 Reactions with dichlorocarbene sources and pentafluorobenzotrichloride. Synthesis of polyfluoroarenes having imidoyl chloride group
Thermolytical reactions of polyfluoroarenes with dichlorocarbene sources were studied by the example of polyfluoroaromatic amines. Chloroform was used as a dichlorocarbene source. 2,3,4,5,6-Pentafluorobenzotrichloride was also introduced in the reaction as a potential source of pentafluorophenylchlorocarbene. Copyrolysis of polyfluoroaromatic amines with these compounds in a flow system at temperatures 500–670 °C occurs involving only the amino group and does not affect the polyfluoroaromatic ring. As a result, the main products of the reaction with chloroform are N-polyfluoroarylcarbonimidoyl dichlorides obtained in 14–34% yield [39,40], and the reaction with pentafluorobenzotrichloride gives N-(polyfluoroaryl)pentafluorobenzimidoyl chlorides in 50–77% yield [41].
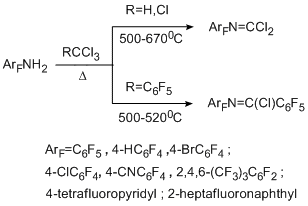
In the reaction with 4-bromotetrafluoroaniline, the exchange of bromine atom for chlorine takes place, and 4-chloroimidoyl chloride derivative proved to be the final product. The attempt to involve non-fluorinated amines, for example aniline, in these transformations failed, probably, because of its thermal instability. At the same time, the method proved to be sufficiently general for obtaining polyfluoroarylimidoyl chlorides, and it is applicable not only to polyfluorinated amines of the benzene series, but also to polyfluorinated amines of the naphthalene and pyridine series.
The possible routes for the formation of imidoyl chloride derivatives are shown in the following scheme with an example of the reactions with chloroform:
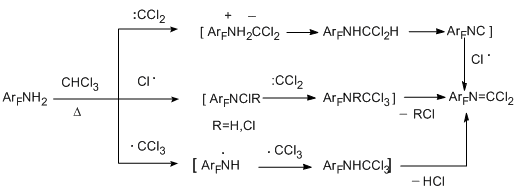
In the case of chloroform, the reaction route involving trichloromethyl radicals should not also be excluded, although the main route of thermal decomposition of chloroform results in dichlorocarbene. Actually, the reaction with carbon tetrachloride as a notorious source of trichloromethyl radical, leads to the same carbonimidoyl dichloride derivatives even in the higher yield (21 to 37%) because of decreasing the content of by-products in the reaction mixture [40]. Most probably, the reaction with pentafluorobenzotrichloride also proceeds via radical mechanism.
3 Radical reactions
Thermolytic radical transformations of aromatic compounds and their polyfluorinated analogues proved to be promising for obtaining polyfluorinated heterocyclic compounds. They are based on the assumption that the starting arenes (ArXR and ArFXR), containing a functional group with a heteroatom (X) at the benzylic position, give radicals of benzyl-type (ArX· and ArFX·) or aryl-type (Ar· and ArF·). These radicals are supposed to react with polyfluoroolefins, and the following intramolecular cyclization takes place to form a heterocyclic system.
3.1 Reactions involving benzenethiyl radicals. Copyrolysis of aryl thiols with polyfluorinated olefins in the presence of oxidizing agents. Synthesis of polyfluorinated dihydrobenzothiophenes
The copyrolysis of aryl thiolsor their polyfluorinated analogues with tetrafluoroethylene or chlorotrifluoroethylene in the presence of oxidizing agents can serve an example of such reactions. Benzenethiol, diphenyl disulfide and their derivatives were studied as substrates, they easily generate radicals ArS· or ArFS·by the action of oxidizing agents (iodine or its source – 1,2-diiodotetrafluoroethane).
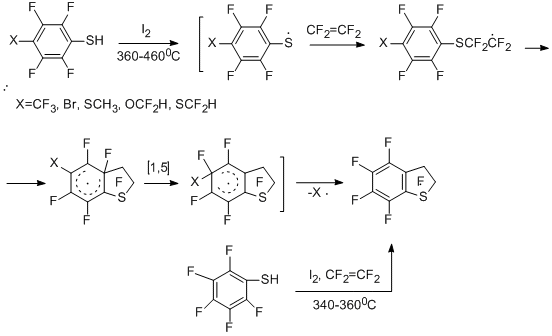
Polyfluorinated 2,3-dihydrobenzo[b]thiophenes are the reaction products [42,43]. Elimination of substituents in para-substituted polyfluorobenzenethiols [43] occurs similarly to the reactions of these compounds with tetrafluoroethylene in the absence of oxidizing agents (see Section II.2.1). The copyrolysis of 2-Н-3,4,5,6-tetrafluorobenzenethiol with tetrafluoroethylene in the presence of either iodine, or 1,2-diiodotetrafluoroethane gives mainly perfluoro-2,3-dihydrobenzo[b]thiophene.
3.2 Reactions involving radicals of the phenyl type
3.2.1 Copyrolysis of polyfluoroaromatic nitro- and sulfonyl chloride derivatives with tetrafluoroethylene. Formation of perfluorotetraline, perfluoroindan, and their heteroanalogues
Copyrolysis of tetrafluoroethylene with с pentafluoronitrobenzene, 4-nitrotetrafluoropyridine and pentafluorobenzenesulfonyl chloride gave very peculiar results. Unlike the above-mentioned similar reactions of other polyfluoroarene derivatives with tetrafluoroethylene as the difluorocarbene source, in this case, the main products proved to be perfluorotetraline and perfluoro-5,6,7,8-tetrahydroisoquinoline [19,25,28], in addition to perfluoroindan and its heteroanalogues.
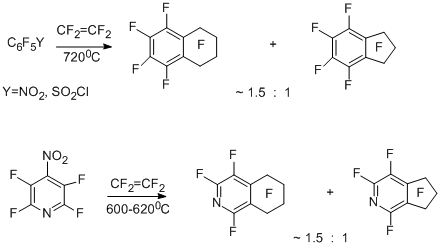
Such direction of the reaction is explained by that the initial nitro- and sulfonyl chloride derivatives generate polyfluorinated aryl radicals under the reaction conditions, these radicals react with tetrafluoroethylene molecules to give perfluoroarylethyl and perfluoroarylbutyl radicals, and the subsequent intramolecular cyclization of them leads to a perfluorinated six-membered rings.
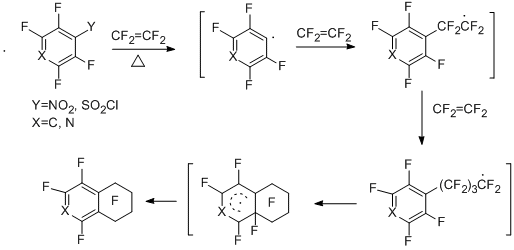
3.2.2 Copyrolisis of polyfluoroaromatic nitro- and sulfonyl chloride derivatives with butadiene. Formation of 1,2,3,4-tetrafluoronaphtalene and 1,3,4-trifluoroisoquinoline
Copyrolysis of pentafluorobenzenesulfonyl chloride, 2,3,5,6-tetrafluoropyridine-4-sulfonyl chloride, and pentafluoronitrobenzene with butadiene leads to 1,2,3,4-tetrafluoronaphtalene in the case of benzene derivatives [44] and 1,3,4-trifluoroisoquinoline in the case of pyridine derivative [45].
The yields of tetrafluoronaphthalene prepared from the sulfonyl chloride derivative and the nitroderivative are 40% and 20%, respectively; trifluoroisoquinoline is obtained in 21% yield (according to GLC data). The scheme of the formation of these compounds includes thermal generation of polyfluoroaryl "A"-type radicals from the starting compounds, interaction of these radicals with butadiene to give allyl radical "B", its intramolecular cyclization and the following transformation of the resulting radical σ-complex to give final products.
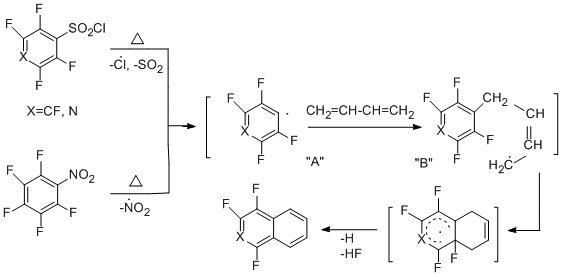
3.2.3 Copyrolysis of polyfluoroarenethiols with chlorine or bromine. Preparation of chloro- and bromopolyfluorobenzenes
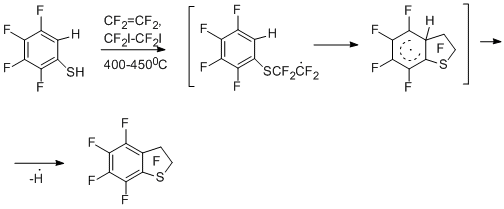
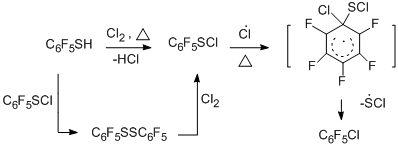
In developing radical thermolytic methods for synthesizing polyfluoroarenes and -hetarenes, a new process for obtaining chloro- and bromopolyfluoroarenes and –hetarenes was proposed by copyrolysis of polyfluoroarene- and polyfluorohetarenethiols with chlorine, its sources, or bromine in a flow system at temperatures of 300 – 650 °C [46]. In this case, replacement of the thiol group by chlorine or bromine takes place. The process occurs with high selectivity and the high yields (68 - 95%) of the obtained halogenopolyfluorinated compounds of high purity (96 – 99%). The optimal temperatures for the reactions with chlorine and bromine are 400 and 500 °C, respectively.
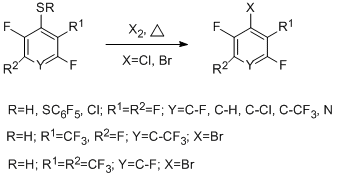
The reaction readily occurs both with pentafluorobenzenethiol and 4-tetrafluoropyridine thiol (400-500 °С) and with 4-substituted 2,3,5,6-tetrafluorobenzenethiols, decafluorodiphenyl disulfide, 2,4- and 2,5-bis(trifluoromethyl)trifluorobenzenethiols, 5-nonafluoroindan thiol. The scheme of formation of halogenopolyfluoroarenes and – hetarenes is presented below by the example of reacting pentafluorobenzenethiol with chlorine, and includes initial transformation of the thiol into sulphenyl chloride, formation of an intermediate radical σ-complex and its transformation in the final product.
Polyfluoroarenes and Zn: hydrogenolysis of perfluoro- and chloropolyfluoroarenes by the action of Zn(Cu)-DMF-H2O, preparation of hydropolyfluoroarenes. Polyfluoroaromatic organozinc compounds
The thermolytic reactions of diverse types considered in Section II, made it possible to obtain a large variety of perfluorinated arenes, hetarenes, and their derivatives. However, as a rule, such reactions are less suitable for synthesizing non-fully fluorinated arenes, because the thermal stability of them or their precursors is lowered. A decrease in thermal stability becomes especially marked, as the number of hydrogen atoms in a molecule increases. For this reason, to obtain compounds having less fluorine atoms than their perfluorinated analogues, hydrodefluorination of the latter by the action of reductive system Zn(Cu)-DMF-H2O has been proposed [47,48]. This reagent leads to hydrogenolysis of the С–F bonds both at the benzylic position and in the aromatic ring. Addition of NaCl, NH4Cl and other salts as electrolytes facilitates hydrogenolysis. The hydrogenolysis does not occur in the absence of water.
The high selectivity is observed in series of perfluoromonoalkylbenzenes, containing perfluorinated methyl, ethyl, propyl, and isopropyl groups [47, 49]. The reaction involves only the C–F bond of aromatic ring at para-position towards the perfluoroalkyl group. The proposed mechanism of hydrogenolysis of the CAr –F bond includes the formation of intermediate anion radical which decomposes to give perfluoroaryl radical and fluoride-ion. The aryl radical adds an electron to form an anion, which reacts with water to give a hydroderivative [47].
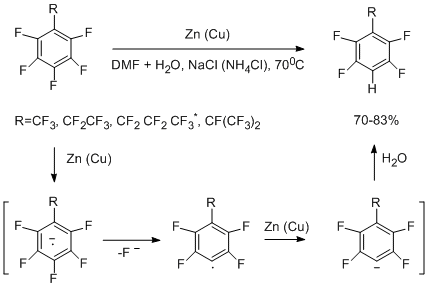
* in the abcence of electrolyte
In accordance with quantum-chemical calculations for anion radicals resulted from octafluorotoluene and perfluoroxylenes [48], realization of the observed direction of the reaction is connected with localization of a free electron in anion radicals at para-position towards to the substituent, this leads to reductive transformation at this particular position and obtaining p-H-substituted perfluoroalkylbenzenes. The hydrogenolysis in perfluoro-p-xylene mainly occurs at the benzylic position with only one CF3-group being involved. The content of 1,4-bis(trifluoromethyl)-3,5,6-trifluorobenzene was small in this case [48].
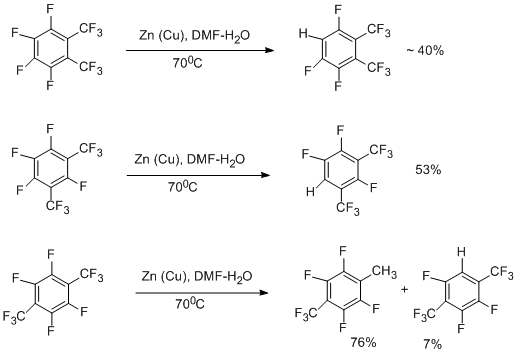
A possible mechanism for hydrogenolysis of benzylic C–F bond assumes that an electron transfer from the metal surface to a perfluoroarene molecule takes place to form anion radical, which undergoes decomposition.
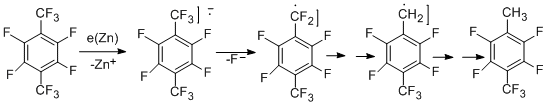
The reaction occurs by steps to form successively groups –CHF2, -CH2F, and finally, CH3. The yield of compounds with CH3-group can achieve 76%, while the content of other products is small.
Perfluoro-p-cymene reacts involving only perfluoroisopropyl group [48].
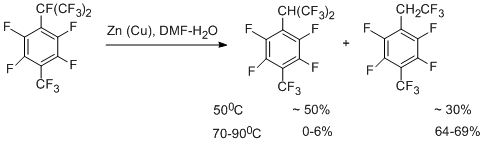
A mechanism for hydrogenolysis of the CАlk-F bond that results in formation of hydrogenized hexafluoroisopropyl derivative is similar to that described above for perfluoro-p-xylene, but in this case there are two possible routes for decomposition of the above anion radical, namely, leading to formation of either radical "A", or "B".
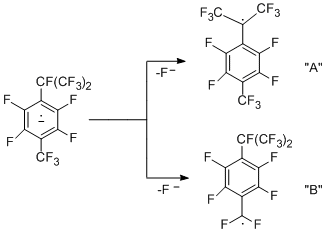
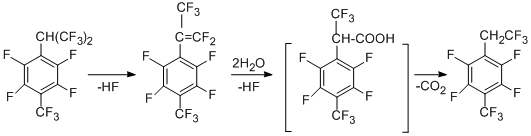
The higher stability of radical "A" compared to that of radical "B" (according to quantum-chemical calculations) and the lower C-F bond strength at the benzylic position of the isopropyl group can be one of the main factors that determine involving only the perfluoroisopropyl group in the reaction. The compound containing trifluoroethyl group, is the product of further transformations of the hexafluoroisopropyl group and is obtained in aqueous DMF both in the presence and in the absence of metals. Its formation can be presented as a result of dehydrofluorination to give perfluoro-a,4-dimethylstyrene, which then reacts with water to afford the corresponding acid. Decarboxylation of the acid gives 1-trifluoromethyl-2,3,5,6-tetrafluoro-4-(2',2',2'-trifluoroethyl)benzene. This scheme is confirmed by transformation of perfluoro-a,4-dimethylstyrene into this ethyl derivative.
The reaction of perfluoro-4-tert-butyltoluene with Zn(Cu)-DMF-H2O in the presence of NaCl occurs in a different manner [49]. In this case, dehydrofluorination takes place mainly at ortho-position towards the bulk perfluoro-tert-butyl group.
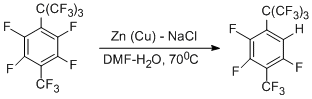
Hydrodefluorination of some perfluoroarenes can occur in an aqueous medium in the presence of electrolytes without addition of DMF. Under these conditions, replacement of the fluorine at position 4 by hydrogen takes place, for example, in octafluorotoluene and pentafluoropyridine [50], although in this case some amount of starting arene remains unreacted.
When an alkyl group in a perfluoroarene contains chlorine or bromine atoms, hydrogenolysis proceeds involving exclusively these atoms [51]. Only in the case of 4-chloro-2,3,5,6-tetrafluorobenzotrichloride, the product of hydrogenolysis at the CAr-Cl bond was obtained (13%), in addition to the product of the major reaction at benzylic position (yield 78%).
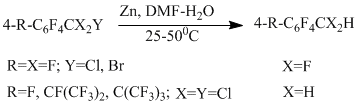
Generally, hydrogenolysis of a CAr-Cl bond is the only direction of the reaction in chloropolyfluorobenzenes (chloropentafluorobenzene, a mixture of isomer dichlorotetrafluorobenzenes, 1,3,5-trichlorotrifluorobenzene) [51], this is connected with the structure of the intermediate anion radical of the σ-type, in which an electron interacts with MO localized on the CAr-Cl bond.
The reactivity of C–Hal bond in the considered chloropolyfluoroarenes corresponds to the following regularities: Cbenzyl –Cl > CAr – Cl ; Cbenzyl– Cl > CAr – F; Cbenzyl– Cl > Cbenzyl– F; Cbenzyl– F < CAr – F (para-position).
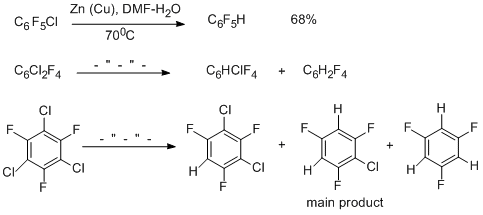
Chloropolyfluoroarenes react with Zn in DMF when the reaction mixture is either heated or treated with ultrasound, to give stable organozinc compounds, which are identified by spectral methods. When they are treated with an acid, they are transformed to give hydro derivatives [51-53]. Only chlorine atoms are involved in the reaction.
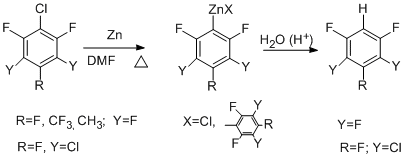
3-Chloroheptafluorotoluene reacts in a similar manner. Addition of SnCl2 to the system favours the formation of organozinc compounds at the expense of involving the CAr–F bond in the reaction [54].
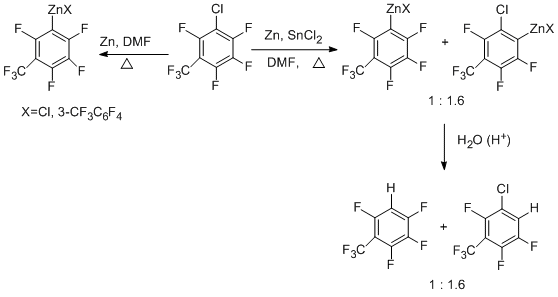
Perfluoroarenes react with Zn–DMF–SnCl2 system with the CAr – F bond being involved to give organozinc compounds even at room temperature [55]. The use of ultrasound favours the reaction. It should be noted that previously the direct formation of CAr – Zn bond with the participation of the CAr – F bond has not been recorded in polyfluoroarenes. The order of substrate activity under the action of Zn–SnCl2 and ultrasound has been found out [55].
Perfluoroindan forms the organozinc compound at position 5 [53].
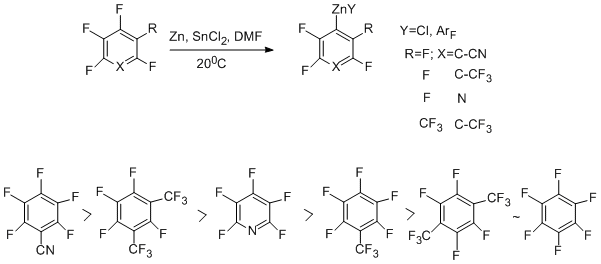
The scheme of formation of organozinc compounds involving C–Cl and CAr –F bonds is presented by the example of the reaction of 3-chloroheptafluorotoluene [54, 56]. In the first case an electron appears to enter into the σ-antibonding orbital of the C-Cl bond (route "A"), whereas the C-F bond is affected as a result of the nucleophilic mechanism contribution (route "B").
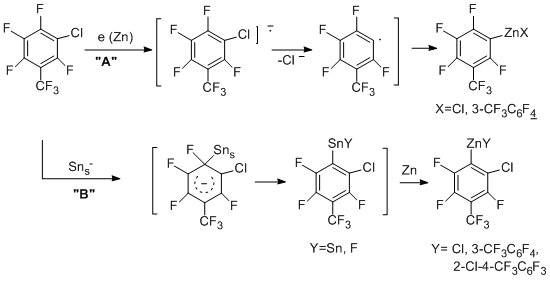
The organozinc compounds obtained from perfluoroarenes are converted to hydro-, bromo-, iodoperfluoroarenes and perfluorobiaryls under the action of H2O (H+), Br2, I2 and CuCl2, respectively [55]. The reactions with allyl chloride in the presence of CuCl as a catalyst or allyl bromide give allylperfluoroarenes [53]. The reactions with allyl chloride in the presence of CuCl appears to occur via polyfluoroaryl organocopper compounds.
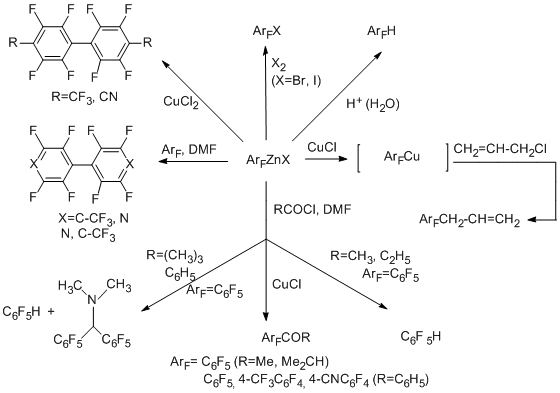
Polyfluorobiaryls are resulted from the reacting the polyfluoroaryl organozinc reagent as a nucleophile with polyfluoroarenes. The product of reactions with acid chlorides depends on the nature of an acid and the reaction conditions. Such products can be either ketone [57], or a mixture of hydroperfluoroarene and a substituted amine, which is obtained as a result of involving a Vilsmeier-Haack-type reagent in the reaction. The similar transformations are also realized for fluorinated organozinc compounds of the pyridine and indan series.
Chemistry of functional derivatives of polyfluoroarenes
1 Intramolecular nucleophilic substitution of ortho-fluorine in polyfluoroarenes. Synthesis of heterocyclic and carbocyclic systems
Nucleophilic substitution of fluorine atoms is the most typical transformation of polyfluoroarenes. Substituted polyfluoroarenes having a substituent of the corresponding structure with a nucleophilic reaction centre in it, can undergo reactions of intramolecular nucleophilic substitution with elimination of the ortho-fluorine to form either heterocyclic or carbocyclic ring. This transformation became the basis for a general and convenient method for synthesizing polyfluorinated benzheterocyclic compounds that made it possible to obtain a wide range of various benzheterocyclic systems including those that are scarcely available by other routes. The reaction can be performed by direct action of a bifunctional nucleophile on a perfluoroarene either with isolating the primary reaction product with one nucleophilic centre followed by cyclization, or without isolating this primary product. The examples are given below [58].
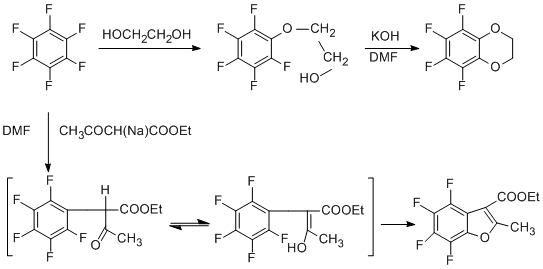
Cyclization of pentafluorophenetylamine [59], isomeric trifluorophenetylamines [60] and β-pentafluorophenylpropylamine [61] under the action of KF in DMF gives polyfluorinated indoline derivatives. The KF/DMF system is often used in reactions of intramolecular nucleophilic substitution.
When a substituent in a perfluoroarene contains two nucleophilic centres, the interesting results are obtained. For instance, cyclization of α-benzamido-β-pentafluorophenylacrylic acid occurs involving only the carboxy group to give a cumarine derivative, whereas in cyclization of methyl ester of this acid the benzamide group participates that behaves as an enol to lead to a benz[f]oxazepine-1,3 derivative [62]. Probably, the nucleophilicity of benzamide group is low, and the presence of an electron-acceptor substituent favours enolization of this group so that the active nucleophilic centre proves to be at the oxygen.

The reaction of intramolecular nucleophilic substitution was also used for the preparation of dibenzosubstituted polyfluorinated heterocycles, for example, phenothiazines [63,64], phenoxazines [63], and other systems [65].
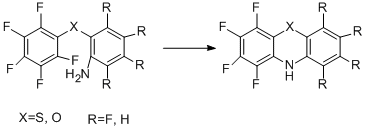
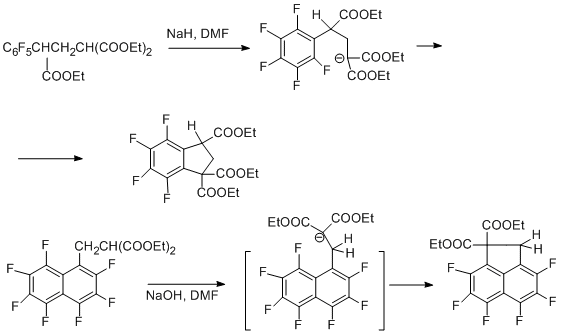
If the nucleophilic centre is on the carbon, intramolecular nucleophilic substitution leads to a carbocyclic system [66].
2 Reactions of intramolecular electrophilic cyclization
Like nucleophilic substitutions as a whole, the above-described intramolecular nucleophilic substitution with eliminating a fluoride anion from the ortho-position is a very distinctive type of transformations for polyfluoroarenes. At the same time, the intramolecular electrophilic closure with eliminating fluorine cation from the ortho-position is not energetically favourable process. However, intramolecular electrophilic substitution proved to be possible, this was shown by the examples of the formation of polyfluorinated carbocyclic compounds. When perfluoro-1-phenyl- and -3-phenylpropenes react with AlCl3 (90 – 100 °C), 1,1,3,3-tetrachlorohexafluoroindan is formed, in addition to 1,1,3,3,5-pentachloropentafluoroindan (15%) and small amount of pentafluorophenyl-2-fluorotetrachloropropene [67].
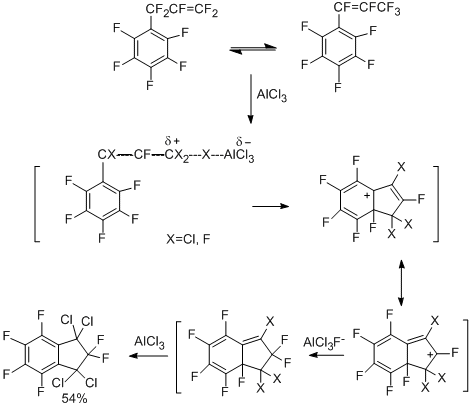
These compounds are also formed when hexafluoropropene is reacted with pentafluorobenzene in the presence of AlCl3, where at first electrophilic halogenoalkenylation appears to take place with substituting the hydrogen by the perhalogenopropenyl group, to result in formation of 1,1,3,3-tetrachlorohexafluoroindan. The process is accompanied by exchange of the fluorine for chlorine. The scheme of cyclization can include activation of 1-pentafluorophenyl-2-fluoropolyhalogenopropenes by the action of AlCl3 followed by electrophilic ortho-addition and moving halogens in the bicyclic system. The formation of 1,1,3,3,5-pentachloropentafluoroindan can occur via replacement of the fluorine by chlorine in 1,1,3,3-tetrachlorohexafluoroindan under the action of AlCl3 [68].
The formation of perfluoro-1,7-dimethylindan, a product of intramolecular electrophilic cyclization, was observed when perfluoro-α, β, o-trimethylstyrene was reacted with SbF5 at 200 °C [69].
3 Polychloroalkylation of polyfluoroarenes having N-, O-, and S-containing functional groups in the presence of AlCl3, affecting heteroatom
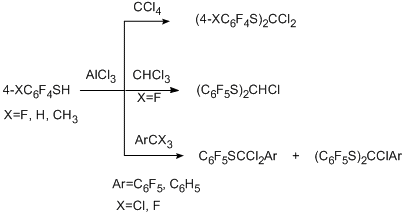
One of the successfully developed methods for transformation of functional groups in polyfluoroarenes is the reaction of electrophilic polychloroalkylation of polyfluorinated aromatic N-, O- and S-containing functional derivatives by the action of system RCX3/AlCl3 (X=Cl, F) that occurs via formation of a polychloroalkyl group at the a-position towards heteroatom. The method is based on the attack at the donor N-, O- or S-heteroatom by an intermediate electrophilic complex, this complex is formed from a trihalogenomethyl reagent by an action of AlCl3. The polychloromethyl derivative that formed can be isolated or it undergoes further transformations to give compounds of the various types. The reaction is sufficiently general [70,71]. As substrates polyfluorinated amines of benzene, naphthalene, diphenyl series [72], arylhydrazines [73] (non-fluorinated aromatic amines and hydrazines with acceptor nitro group also enter this reaction), pentafluorophenol [74], pentafluorobenzaldehyde [75], polyfluorobenzenethiols [76] are used. Compounds RCX3 comprise CCl4, aromatic derivatives containing one or more groups CX3, including poly- and perfluorinated derivatives as well as perhalogenated olefins with terminal group CCl3 (CClX=CYCCl3; X = Cl, C6F5; Y = Cl, F) [77]. Substitution of the hydrogen atoms in aromatic ring of a substrate for fluorine favours the successive development of the method, because this excludes an alternative route of polychloroalkylation at the CAr-H bond.
Polyfluorinated aromatic amines [72] and hydrazines [73] in reactions with RCX3 and AlCl3 give N-trichloromethyl derivatives, under the reaction conditions, they are converted respectively to N–polyfluoroarylcarbonimidoyl dichlorides or N-trichloromethyl-N-polyfluoroarylhydrazonodichloromethanes, when R=X=Cl, or to N-polyfluoroarylimidoyl chlorides and N-polyfluoroarylhydrazonochlorides, when R=Ar, ArF, and X=Cl, F. The obtained carbonimidoyl dichlorides and imidoyl chlorides are identical to those synthesized in the reactions of amines described above (Section II.2.2).
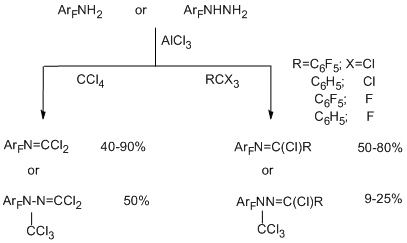
The use of CCl3-substituted olefins as polychloroalkylating reagents [77] makes it possible to obtain polyhalogenated 4-aza-1,3-butadienes.
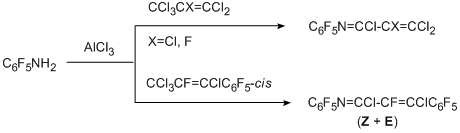
When compounds containing several perhalogenated alkyl groups (isomeric perfluoroxylenes, perfluoro-p-cymene, 1,3-bis(trichloromethyl)tetrafluorobenzene, 4-(trichloromethyl)undecafluoroisopropylbenzene) are used under studied conditions, the formation of the imidoyl chloride moiety occurs involving only one CX3-group, and large amount of by-products are formed [78].
The most probable route for reaction of polyhalogenalkylation is presented by the following scheme [71]. N-polychloroalkylamine formed at the first step of polychloroalkylation can be isolated if its further transformation into an imidoyl chloride derivative is impossible, for example, as in the case of decafluorodiphenylamine and N-methylpentafluoroaniline [72].
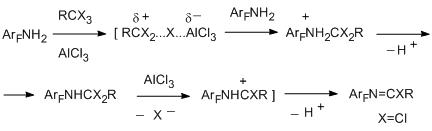
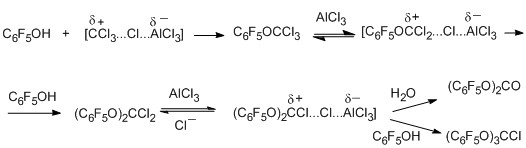
Polychloroalkylation of pentafluorophenol by the action of CCl4 and AlCl3 occurs according to a similar scheme to form the primary polychloroalkylation product, namely, trichloromethylpentafluorophenyl ether, as well as bis(pentafluorophenyl) carbonate as a product of hydrolysis of the formed dichloromethane derivative, and tris(pentafluorophenoxy)chloromethane [74]. Varying the reaction conditions makes it possible to shift the system to the side of the predominant formation of one of the product (for example, up to 40% for the initial ether and more than 45% for the carbonate).
The use of benzotrihalogenides ArCX3 (X=Cl, F; Ar=C6H5, C6F5) instead of CCl4 leads to pentafluorophenyl esters of benzoic acids which are the products of hydrolysis of the intermediate compounds C6F5OCX2 Ar (yield ~ 50%) [79].
Direct polychloroalkylation at the oxygen atom of pentafluorobenzaldehyde gives pentafluorobenzylidene chloride in high yield as the final product [75].
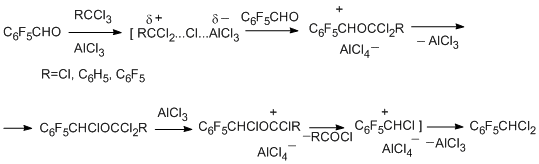
Like polyfluorinated phenols, polyfluorinated benzenethiols undergo polychloroalkylation at the sulfur atom, however, in this case, the primary products of polychloroalkylation have been obtained only with benzotrihalogenides, and the main products of the reaction are polyfluoroaryl sulfides containing a substituted methylene group, which are stable towards hydrolysis [76]. The differences in the results obtained in reactions of polyfluorinated phenols and benzenethiols are explained by the higher nucleophilicity of benzenethiols and the better ability of the oxygen atoms to stabilize the adjacent carbocation centre compared to that of the sulfur.
4 Transformations of polyfluoroarenes containing imidoyl chloride moiety
4.1 Reactions with nucleophilic reagents
Polyfluoroaromatic imidoyl chloride derivatives are highly reactive compounds which can be involved in transformations of various types. The most typical of them are reactions with nucleophilic reagents. They proceed involving the chlorine atoms at the N=C bond and with retention or transformation of this bond. The reaction products are either stable or they undergo further transformations. The acceptor properties of polyfluoroaromatic ring and its ability to serve as an additional reactive centre lead to the secondary transformations, including heterocyclization which is the most interesting of them.
4.1.1 N–Nucleophilic reagents
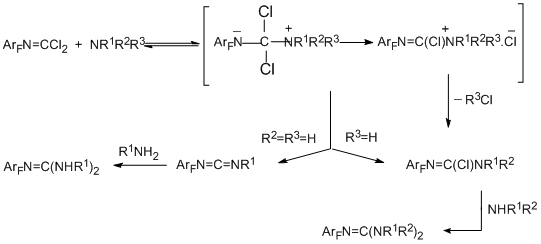
The regularities observed in reactions of polyfluoroaromatic carbonimidoyl dichlorides and imidoyl chlorides with primary, secondary and tertiary amines are similar to those earlier observed in the corresponding reactions of their non-fluorinated analogues. In reactions of carbonimidoyl dichlorides with primary amines, both chlorine atoms are readily eliminated, and the product type depends on the nature and amount of the amine. The reactions with primary aliphatic amines give carbodiimides and guanidines. The reactions with primary aromatic amines give only guanidines. In the case of secondary amines independently on their nature, at first elimination of one chlorine atom and formation of chloroformamidines occur, and then the second chlorine atom is eliminated to give guanidines [80,81].
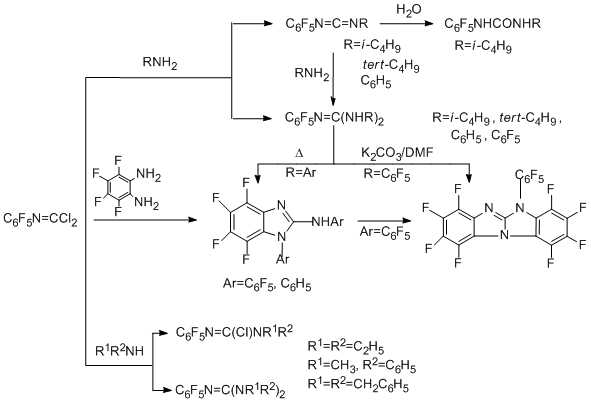
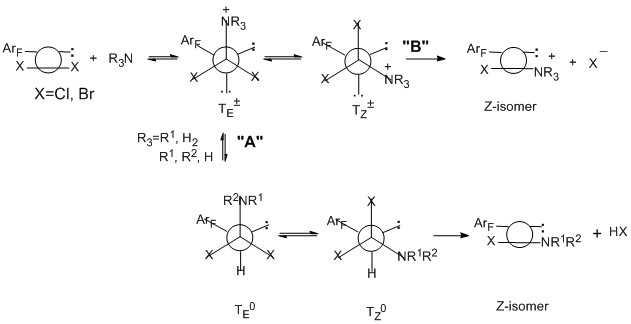
Polyfluoroaromatic imidoyl chloride derivatives reacting with tertiary amines give imidoylammonium salts, which are stable in the case of aromatic amines (for example, 4-N,N-dimethylaminоpyridine) and can be isolated in quantitative yield [82], whereas in the case of aliphatic amines, they are formed as intermediate compounds which are converted to chloroformamidines [83]. Reactions with all amines occur by a bimolecular addition-elimination mechanism to form a tetrahedral intermediate ТЕ± at the first step [83, 84]. In the case of primary and secondary amines, the formation of this intermediate is a rate-limiting step followed by transformation resulting in Z-isomer of chloroformamidine (route "A"). A rate-limiting step for the reactions with tertiary amines is a stereo mutation of the primary tetrahedral intermediate TЕ± to give thermodynamically more stable TZ± and then a salt is formed (route "B").
The following scheme explains the formation of different products in the case of primary and secondary amines (carbodiimides/guanidines and chloroformamidines, respectively):
Chloroformamidines and guanidines are also obtained if reactions of polyfluoroarylimidoyl dichloride derivatives with aromatic amines are carried out in the presence of AlCl3 [85]. When N-pentafluorophenylcarbonimidoyl dichloride is reacted with amines and their polyfluorinated in the ring analogues in the presence of AlCl3, guanidines and chloroformamidines are formed under the milder conditions and with the higher conversion degree. This is explained by the different mechanisms of transformations: a bimolecular addition–elimination in the absence of AlCl3 and a mechanism including polarization of an imidoyl chloride derivative by the action of AlCl3 resulting in a positively charged intermediate, which attacks the nitrogen of an amine. The mechanism of such a type will be considered in detail in Section 4.2.
4.1.2 O-Nucleophilic reagents
The reactions of N-pentafluorophenylcarbonimidoyl dichloride with alcohols, phenols, and ethylene glycol have been studied in detail [86]. In the absence of bases, addition of a nucleophile to the C=N bond occurs to form pentafluorophenyl carbamates as the final products. In the presence of a base, for example, K2CO3, one or both chlorine atoms are eliminated, the С=N bond remains, and either mono- or diimidates are formed, or intramolecular heterocyclization can occur as in the case of ethylene glycol.
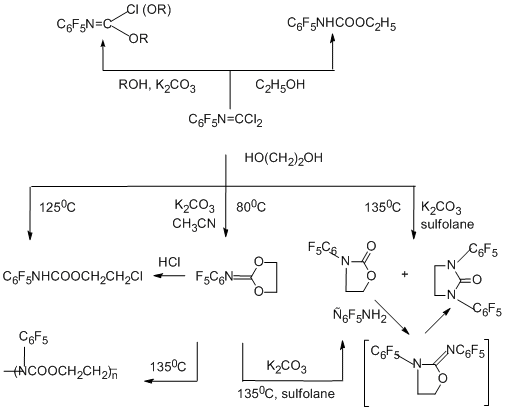
4.1.3 S-Nucleophilic reagents
Reactions of polyfluoroaryl imidoyl chloride derivatives with benzenethiols, thiourea, sodium N,N-diethyldithiocarbamate and –SCF3 anion sources have been studied [87], but only the first three reagents are synthetically interesting.
The main direction of the reactions with benzenethiols in acetonitrile and in the presence of anhydrous K2CO3 is substitution of one or two chlorine atoms for the benzenethiol moiety. When the C6F5 group is present at the carbon atom of the N=C multiple bond, preferential substitution of the para-fluorine atom in this group occurs. This is connected with that according to quantum-chemical calculations for imidoyl chloride derivatives of such a type, for example, N-pentafluorophenyl(pentafluorobenz)imidoyl chloride, the reaction pathway is determined by orbital control, and the largest frontier densities of LUMO are concentrated at the para-positions of C-pentafluorophenyl ring and at the carbon atom of the N=C bond [87]. For the analogue having phenyl group at the C-atom and for N-pentafluorophenylcarbonimidoyl dichloride, the reaction pathway is determined by the gain in energy of formation of the intermediate σ-complex on addition of S-nucleophile to the carbon atom of the N=C multiple bond.
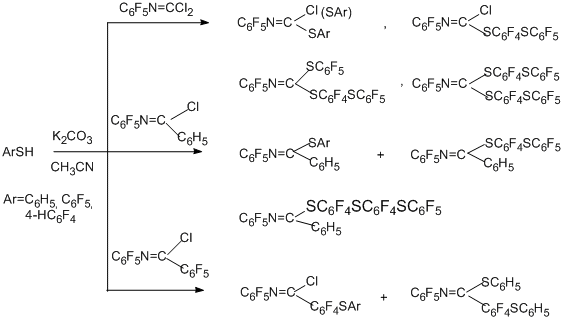
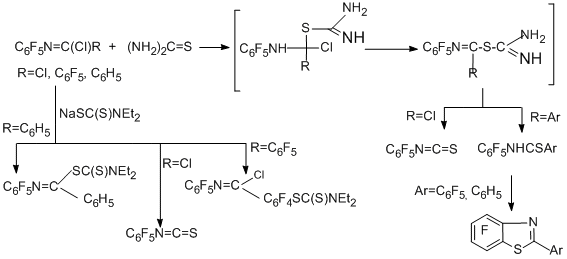
Thiourea and sodium-N,N-diethyldithiocarbamate give either pentafluorophenyl isothiocyanate, or heterocyclic derivatives [87].
4.1.4 Halogenide-anions
Successive substitution of chlorine for bromine is observed in reaction of N-pentafluorophenylcarbonimidoyl dichloride by the action of either LiBr in acetonitrile, or AlBr3 in dibromomethane [79].
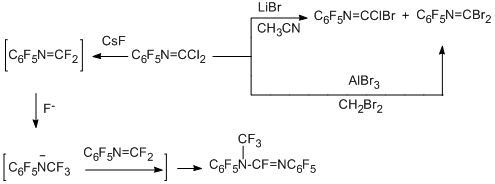
The attempts to substitute the chlorine by fluorine and isolate carbonimidoyl difluoride by the action of CsF under various conditions gave only dimer of this difluoride [79].
4.2 Reactions in the presence of AlCl3
Polyfluoroarenes containing imidoyl chloride moiety behave as effective electrophiles in the presence of AlCl3, this makes it possible to introduce reactive groups ArFN=CCl- or ArFFN=CАr- into substrates. In the course of the transformations, cycloaddition or intramolecular cyclization can take place which lead to polyfluorinated N-heterocyclic compounds.
Reactions of N-pentafluorophenylcarbonimidoyl dichloride with aromatic hydrocarbons (benzene, toluene, xylene isomers, mesitylene) occur under mild conditions to give stable imidoyl chlorides, which are converted to azomethines [88]. Predominant para-orientation relative to the СН3-group is observed at both steps of the reaction. The reactivity of hydrocarbons increases in the following order: benzene < toluene < para-xylene < ortho-xylene < meta-xylene < mesitylene. Less basic fluorobenzene and 1,3,5-trifluorobenzene react in a similar manner, but under more severe conditions [89]. The regularities observed in reactions of arenes and fluoroarenes make it possible to conclude that the reactions have electrophilic character and include polarization of the imidoyl chloride derivative under the actions of AlCl3 with the formation of an intermediate nitrilium-type cation followed by the attack at the hydrocarbon by the positively charged centre of the intermediate.
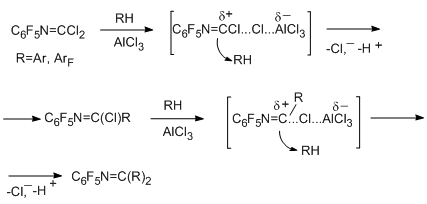
In the case of 1,2,4,5-tetrafluorobenzene and pentafluorobenzene, intramolecular cyclization occurs preferably at elevated temperature to give polyfluorinated 1,2,3,4-tetrahydroquinazolin-2,4-diones. Replacement of fluorine for chlorine is a side reaction [89]. These data indicate that accumulation of fluorine atoms in the aromatic ring leads to changes in reactivity of the compound, and the frontier-line is observed when passing from three to four fluorine atoms.
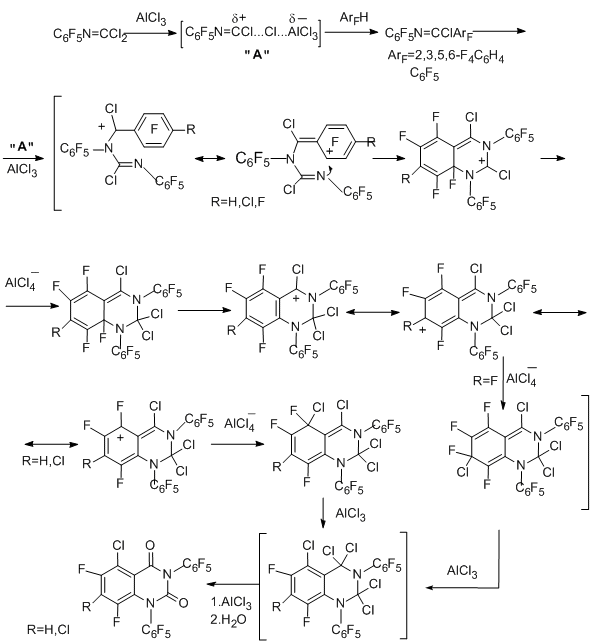
Probably, reactions of polyfluorinated imidoyl chloride derivatives with compounds containing nitrogen–carbon multiple bonds in the presence of AlCl3 occur involving a cationoid intermediate of the nitrilium-type cation to form products of "cycloaddition", namely, five- and six-membered N-heterocyclic compounds. These reactions can be considered as a new route to polyfluorinated N-heterocyclic systems. For example, when 4-R-tetrafluorophenycarbonimidoyl dichlorides are reacted with N-pentafluorophenyltrichloroacetimidoyl chloride (N=C multiple bond), 2-imidazolidone derivatives are formed, and "dimerization" of the mentioned trichloroacetimidoyl chloride in the presence of AlCl3 gives substituted 2-imidazolone [90].
The distinction in the products of the "dimerization" and the reaction between the two different imidoyl chloride derivatives can be explained by the higher stability and the easier formation of the cationoid intermediates in the case when X = Cl in comparison with the case, when X = CCl3.
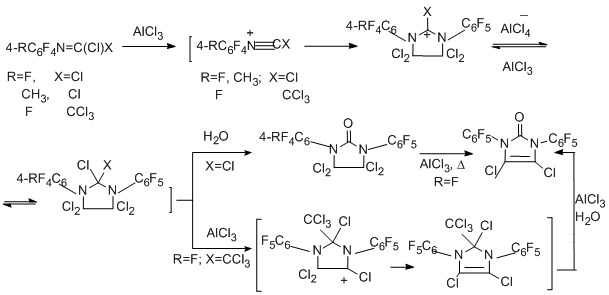
Substituted triazine has been obtained in the reaction with benzonitrile (N≡C multiple bond) at high temperature [90].
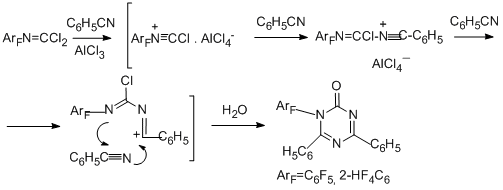
Finally, reactions of N-pentafluorophenylcarbonimidoyl dichloride with benzoic and phthalic acids or their derivatives (acid chlorides, ethyl benzoate, phthalic anhydride) at 170°С in the presence of AlCl3 lead to 2-(pentafluorophenyl)isoindolin-1,3-dione as the main product; in the case of phthalic acid and its derivatives the yield of the product is more than 90% [91].
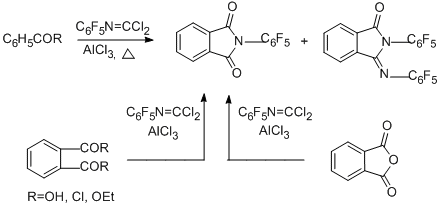
2-(Pentafluorophenyl)-3-[(pentafluorophenyl)imino]isoindolin-1-one is the second main product in the reaction with benzoyl chloride. In the case of benzoic acid, the main product is the benzoyl derivative of pentafluoroaniline. The possible routes for formation of the compounds observed in the reaction with benzoyl chloride are presented in the following scheme.
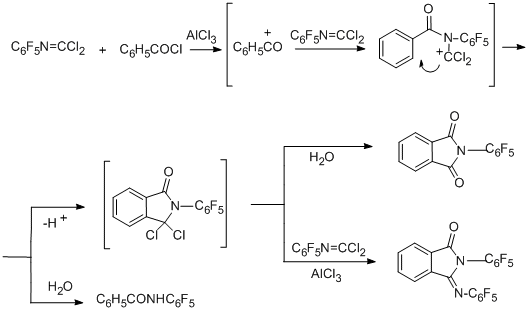
The reactions of polyfluoroaromatic imidoyl chloride derivatives with aromatic amines in the presence of AlCl3 were reported above (see Section IV.4.1.1).
5 Nitration and nitrosation of polyfluoroaromatic N-alkylamines. Preparation of N-nitro- and N-nitroso-N-alkylpolyfluoroarylamines
Nitration and nitrosation of polyfluoroaromatic N-alkylamines is advancing the methods for transformation of functional groups in polyfluoroarenes under the action of electrophilic reagents.
Interaction of N-alkylpolyfluoroarylamines with nitric acid gives N-nitro-N-alkyl derivatives [92]. In some cases N-nitrosо compounds are obtained as minor products. The reaction is sufficiently general and occurs satisfactorily even at lowered basicity of the amine at the expense of introducing additional acceptor groups or using bulk alkyl groups. In the case of tert-butyl group, N-nitroamines are slightly stable, and their formation can be confirmed only by spectral data.
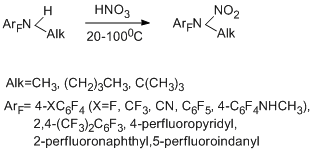
A mechanism of nitration includes an electrophilic attack at the nitrogen atom of the amino group by a nitronium cation generated from nitric acid. Involving the nitronium cation in the reaction is confirmed by the formation of the same N-nitroalkylamines when the notorious source of such cation – nitronium tetrafluoroborate – in sulfolane was used. It is true that in this case N-nitroso compounds are obtained as minor products.
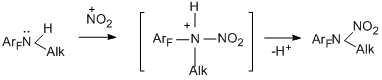
By the example of reactions of N-methylpentafluoroaniline and N-methylperfluoro-p-toluidine with a mixture of nitric and sulfuric acids, the formation of tetrafluoro-p-benzoquinone and heptafluoro-p-toluquinone, respectively, as another type of reaction products has been found. This can be explained by a decreased basicity of the nitrogen of the amino group because of protonation, and as a result, the attack of the nitronium cation is directed at the carbon atom of aromatic ring [92].
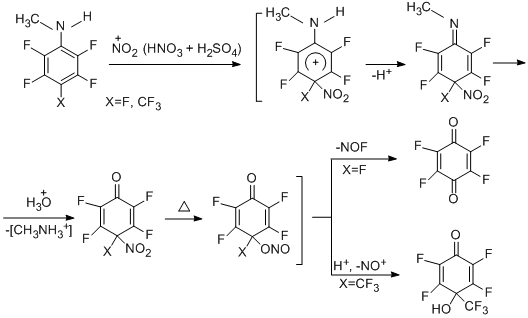
The reactions of polyfluoroaromatic N,N-dimethylamines with nitric acid and with a mixture HNO3 + H2SO4 occur with conversion of the side chain to give N-nitroso derivatives as the main products in the form of a mixture of Е and Z-isomers with the E-isomer predominating (~2.2–3 : 1) [93].
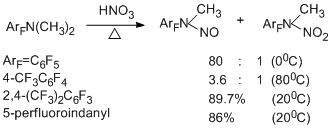
N-Nitroso derivatives are converted to the corresponding N-nitro derivatives as a result of action of a mixture of HNO3 and H2SO4, HNO3 or NO2BF4 [93]. The scheme given below shows the possible routes for the formation of N-nitroso- and N-nitroalkylamines [93].
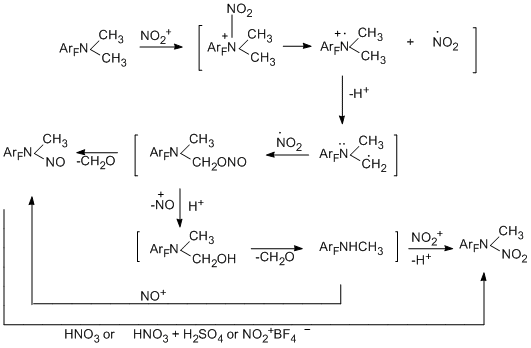
N-Nitroso-N-alkylpolyfluoroarylamines, which could be of interest as biologically active compounds can be obtained in yields from 56 to 94% by nitrosation of N-monoalkylpolyfluoroarylamines under the action of NaNO2 in HCl or CH3COOH [94]. In this case, a mixture of E and Z-isomers is formed, the isomer ratio depends on the nature of the starting amine.
6 Reactions of polyfluoroarenethiols with electrophiles. Synthesis of polyfluoroarenesulfonyl bromides
Another example of electrophilic transformation of a functional group of a polyfluoroarene or- hetarene is the transformation of the thiol group in polyfluorothiols of benzene, biphenyl, indan, and pyridine series into a sulfonyl bromide group by the action Br2– fuming HNO3 or other oxidizing systems (Br2 + HNO3 +H2SO4, HBr + HNO3 + H2SO4, etc.). In most cases the high yields of sulfonyl bromide were achieved (70-91%). In some reactions of pentafluorobenzenethiol the formation of small amount of by-product – decafluorodiphenyl disulfide – was observed; specifically in the case of the use of systems Br2+H2SO4, Br2+CH3COOH, Br2+H2O. By the example of this compound, it was shown that disulfides are also converted to the corresponding sulfonyl bromides under the action of Br2 and fuming HNO3 [95]. Тherefore, a sufficiently simple and convenient method for transformation of a thiol group into a sulfonyl bromide group which is capable of undergoing further transformations including those that give compounds with practically important properties.
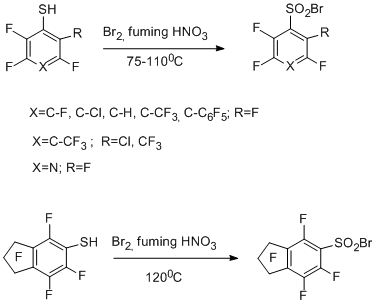
Electrophilic brominating agents and a nitronium cation are supposed to be involved in the formation of sulfonyl bromide group. One of the possible routes of the reactions can include the formation of intermediate ArFSBr as a result of interaction of polyfluoroarenethiol with electrophilic agents (this intermediate can also be responsible for the formation of ArFS-SArF). The following interaction of intermediate ArFSR (R=Br, SArF) with Br2 and ONO2- gives intermediate ArFS(O)Br, which is converted to the end product.
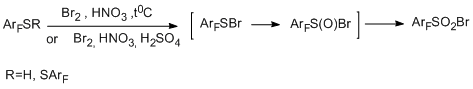
Conclusion
The considered reactions of polyfluoroaromatic compounds have demonstrated new synthetic possibilities in this field of chemistry and led to studying a number of aspects for basic research. Elucidation of new regularities in reactivity of polyfluoroaromatic compounds would favour the development of their synthesis potential. The investigations could be very useful for organic chemistry as a whole. The new discovered synthetic opportunities of polyfluoroarenes could stimulate interest in searching for new fields of their practical applications.
References
- Vorozhtsov-jr. N.N., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1963. N. 8. S.1524
- Vorozhtsov-jr.N.N., Yakobson G.G. // Zhurn. obshch. khimii. 1957. T. 27. Vyp. 6. S. 1672-1676.
- Vorozhtsov-jr. N.N., Yakobson G.G. // Zhurn. obshch. khimii. 1961. T. 31. Vyp. 11. S. 3705-3708.
- Yakobson G.G., Platonov V.E.,Vorozhtsov-jr. N.N. // Zhurn. obshch. khimii. 1965. T. 35. Vyp. 7. S 1158-1161.
- Yakobson G.G., Shteingarts V.D., Vorozhtsov-jr. N.N. // Izv. AN SSSR. Ser. khim. 1964. N 8. S. 1551.
- Yakobson G.G., Vlasov V.M., Vorozhtsov-jr.N.N. // DAN SSSR. 1966. T. 169. N. 4. S. 855-857.
- Yakobson G.G., Odinokov V.N., Petrova T.D., Vorozhtsov-jr. N.N. // Zhurn. obshch. khimii. 1964. T. 34. Vyp .9. S. 2953-2958.
- Yakobson G.G., Odinokov V.N., Vorozhtsov-jr. N.N. // Zhurn. obshch. khimii. 1966. T. 36. Vyp. 1. S. 139-142.
- Vlasov V.M., Odinokov V.N., Rudakova R.I., Yakobson G.G., Vorozhtsov-jr.N.N. //Zhurn. obshch. khimii. 1967. T. 37. Vyp. 1. S. 171-176.
- Yakobson G.G., Shteingarts V.D., Kostina N.G., Osina O.I., Vorozhtsov-jr.N.N. //Zhurn. obshch. khimii. 1966. T. 36. Vyp. 1. S. 142-147.
- Yakobson G.G., Platonov V.E., Kryukova V.S., Gershtejn N.A., Vorozhtsov-jr.N.N. //Zhurn. obshch. khimii. 1966. T. 36. Vyp. 12. S. 2131-2135.
- Yakobson G.G., Petrova T.D., Kobrina L.S. // Fluorine Chem. Rev. 1974.V. 7.P. 115-223.
- Furin G.G., Yumatov V.D. Novoe v khimii poliftoraromaticheskih soedinenij.Novosibirsk : Izd-vo NGPU. 2006. 218 s.
- Vorozhtsov-jr. N.N.,Ezerskij S.N., Kollegov V.F., L'vova A.Ya., Platonov V.E.,Pushkina L.M., Sokolov S.V., Tataurov G.P., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1966.N. 7.S. 1291.
- Vorozhtsov-jr. N.N., Dvornikova K.V., Kollegov V.F., Platonov V.E.,. Pushkina L.M., Sokolov S.V., Tataurov G.P.,Yakobson G.G. // Zhurn. VKhO im. D.I.Mendeleeva. 1969. T. 14.N. 1.S. 114.
- Platonov V.E., Ermolenko N.V., Yakobson G.G., Vorozhtsov-jr.N.N. // Izv. AN SSSR. Ser. khim. 1968. N 12. S. 2752-2754.
- Vorozhtsov-jr N.N., Ermolenko N.V., Mazalov S.A., Osina O.I., Platonov V.E., Tyurin V.S., Yakobson G.G. // Izv.AN SSSR. Ser. khim. 1969. N 1. S. 196.
- Platonov V.E., Ermolenko N.V., Yakobson G.G. // Izv. SO AN SSSR. Ser. khim. nauk. 1978. Vyp. 2. N. 4. S. 117-123.
- Platonov V.E. Diss. dokt. khim. nauk, NIOKh SO RAN. Novosibirsk. 1979.
- Platonov V.E., Ermolenko N.V., Yakobson G.G. // Izv.AN SSSR. Ser. khim. 1970. N12. S. 2843.
- Platonov V.E., Malysheva V.V., Yakobson G.G. // Izv. SO AN SSSR. Ser. khim. nauk. 1977. Vyp. 2. N. 4. S. 133-141.
- Yakobson G.G., Platonov V.E., Ermolenko N.V., Kollegov V.F., Chertok V.S. // Izv. SO AN SSSR. Ser. khim. nauk. 1971. V. 1. N. 2. S.100-102.
- Osina O.I., Chertok V.S., Platonov V.E., Yakobson G.G. // Izv. SO AN SSSR, Ser. khim. nauk. 1978. Vyp. 6. N. 14. S. 138-141.
- Petrova T.D.,Platonov V.E., Gorfinkel' M.I., Yakobson G.G. // Zhurn. organ. khimii. 1975. T. 11. N. 10. S. 2123-2129.
- Platonov V.E., Furin G.G., Malyuta N.G., Yakobson G.G. // Zhurn. organ. khimii. 1972. T. 8. N. 2. S. 430-431.
- Malyuta N.G., Platonov V.E., Furin G.G., Yakobson G.G. // Tetrahedron. 1975. V. 31.N. 9. P. 1201-1207.
- Platonov V.E., Gatilova V.P., Dvornikova K.V., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1974. N. 7. S. 1668-1669.
- Yakobson G.G., Platonov V.E., Furin G.G., Malyuta N.G., Ermolenko N.V. // Izv. AN SSSR. Ser. khim. 1971. N. 11. S. 2615.
- Platonov V.E., Yakobson G.G. // Synthesis.1976. N. 6. P. 374-384.
- Maksimov A.M., Motorina E.E., Platonov V.E., Yakobson G.G. // III Vsesoyuznaya konferenciya pokhimii ftororganicheskih soedinenij, OdessaUSSR. 1978. Tezisy dokladov, S.73.
- Platonov V.E., Maksimov A.M., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1977. N. 10. S. 2387-2388.
- Maksimov A.M., Platonov V.E., YAkobson G.G., Deryagina E.N., Voronkov M.G. //ZHurn. organ. himii.1979.T.15. Vyp. 9. S. 1839-1843.
- Ivanova E.P., Karpov V.M., Platonov V.E., Tataurov G.P., Yakobson G.G.,Yahlakova O.M. // Izv. AN SSSR. Ser.khim. 1972. N. 3. S. 733.
- Maksimov A.M., Platonov V.E., Yakobson G.G., Yahlakova O.M., Bekker R.A., DyatkinB.L., KnunyantsI.L. // Izv. SO AN SSSR. Ser.khim. nauk.1981. Vyp. 6. N. 6. S. 128-133.
- Platonov V.E., Yakobson G.G. // Soviet Scientific Revievs. Section B. Chemistry Reviews. Ed. Vol’pin M.E. Amsterdam: Harwood Acad.Publishers. 1984. V. 5. P. 297-345.
- DvornikovaK.V.,PlatonovV.E.,YakobsonG.G. //Zhurn.organ.khimii. 1975.T. 11. V. 11. S. 2383-2387.
- Dvornikova K.V., Platonov V.E., Yakobson G.G. // Izv. AN SSSR. Ser. khim. 1978. N. 5. S. 1223.
- DvornikovaK.V.,PlatonovV.E.,Yakobson G.G. // J. Fluorine Chem. 1985. V. 28. N.1-2. P. 99-113.
- Savchenko T.I., Petrova T.D., Platonov V.E., Yakobson G.G. // J. Fluorine Chem.1977.V. 9. N. 6. P. 505-508.
- SavchenkoT.I.,PetrovaT.D.,PlatonovV.E.,YakobsonG.G. //Zhurn.organ.khimii. 1979.T. 15.Vyp. 5.S. 1018-1024.
- SavchenkoT.I., Petrova T.D., Platonov V.E., Yakobson G.G. // Zhurn. organ. khimii.1979. T. 15.Vyp. 5.S. 1025-1029.
- Maksimov A.M., Platonov V.E. // Heteroatom. Chem. 1992. V. 3. N. 4. P. 373-384.
- Platonov V.E., Maksimov A.M., Maslovsky P.I. // J. Fluorine Chem. 1995.V. 75. Is. 1.P. 41-49.
- Platonov V.E., OsinaО.I., Maksimov A.M., Kolechkina V.G. // J. Fluorine Chem. 1999. V. 96. N. 2. P. 191-192.
- Kolechkina V.G., Maksimov A.M., Platonov V.E., Osina O.I. // Izv. AN. Ser. khim. 2001. N. 2. S. 307-309.
- Platonov V.E., Maksimov A.M., Dvornikova K.V., Nikul'shin P.V. // Zhurn. organ. khimii. 2005. T. 41. Vyp. 11. S. 1681-1687.
- Krasnov V.I., Platonov V.E. // Zhurn. organ. khimii. 1993. T. 29. Vyp. 5. S. 1078-1079.
- Krasnov V.I., Platonov V.E., Beregovaya I.V., Shchegoleva L.N.// Tetrahedron. 1997. V. 53. Is. 5. P. 1797-1812.
- Krasnov V.I., Platonov V.E. // Zhurn. organ. khimii. 2001. T. 37. V. 4. S. 552-557.
- Krasnov V.I. Diss. kand. khim. nauk. NIOKh SO RAN. Novosibirsk. 1999.
- Krasnov V.I., Platonov V.E. //J. Fluorine Chem. 1992.V. 58. N. 2-3. P. 246.
- Krasnov V.I., Platonov V.E.// Zhurn. organ. khimii. 2000. T. 36. V. 10. S. 1524-1534.
- Vinogradov A.S., Krasnov V.I., Platonov V.E. // Zhurn. organ. khimii. 2008. T. 44. V. 1. S. 101-107.
- Krasnov V.I., Vinogradov A.S., Platonov V.E. // Mendeleev Commun. 2006. N. 3. P. 168-170.
- Miller A.O., Krasnov V.I., Peters D., Platonov V.E., Miethchen R. // Tetrahedron Lett.2000.V. 41. P. 3817-3819.
- Platonov V.E. // 15thEuropean Symposium on Fluorine Chemistry, Prague (CzechRepublic).2007. Book of Abstrs. Р. 157.
- Vinogradov A.S., Krasnov V.I., Platonov V.E. // Mendeleev Commun.2008. V. 18.N.4. P. 227-228.
- Yakobson G.G., Petrova T.D., Kann L.I., Savchenko T.I., Petrov A.K., Vorozhtsov-jr. N.N. // DAN SSSR. 1964. T. 158. N. 4. S. 926-928.
- Petrov V.P., Barhash V.A., Shchegoleva G.S., Petrova T.D., Savchenko T.I., Yakobson G.G. // DAN SSSR. 1968. T.178. N. 4. S. 864-867.
- Petrova T.D., Savchenko T.I., Kukovinec O.S., Yakobson G G. // Izv. SO AN SSSR. Ser. khim. nauk. 1974. V. 2. S. 117-123.
- Petrova T.D., Savchenko T.I., Ardyukova T.F., Yakobson G.G. // Izv.SOAN SSSR.Ser.khim. nauk. 1970. V.3.N. 7.S.119-122.
- Petrova T.D. , Mamaev V.P., Yakobson G.G., Vorozhtsov-jr.N.N. // Khimiya get. soed. 1968. N. 5. S. 771-776.
- Yakobson G.G., Furin G.G., Kobrina L.S., Vorozhtsov-jr. N.N. // Zhurn. obshch. khimii. 1967. T. 37. N. 6. S. 1285-1289.
- Yakobson G.G., Furin G.G., Kobrina L.S., Vorozhtsov-jr. N.N. // Zhurn. obshch. khimii. 1967. T. 37. N. 6. S. 1289-1293.
- Brooke G.М. // J. Fluorine Chem. 1997. V. 86. Is. 1. P. 1-76.
- Vlasov V.M., Petrova T.D., Yakobson G.G. // J. Fluorine Chem. 1972/1973.V. 2. N. 4.P. 373-380.
- Platonov V.E., Dvornikova K.V. // Izv. AN SSSR. Ser. khim. 1989. N 11. S. 2854-2857.
- Karpov V.M., Platonov V.E., Chujkov I.P., Yakobson G.G. // Zhurn. organ. khimii. 1983. T. 19. V. 10. S. 2164-2173.
- Karpov V.M., Mezhenkova T.V., Platonov V.E. // Izv. AN SSSR. Ser. khim. 1990. N 3. S 645-652.
- Petrova T.D., Platonov V.E. // Khimiya v interesah ustojch. razvit. 2007. T. 15. N. 5. S. 511-523.
- Petrova T.D., Platonov V.E. // J. Fluorine Chem. 2005. V. 126. Is. 6. P. 860-876.
- Savchenko T.I., Kolesnikova I.V., Petrova T.D., Platonov V.E. // J. Fluorine Chem.1983.V. 22 N. 5. P. 439-458.
- Petrova T.D., Ryabichev A.G., Savchenko T.I., Platonov V.E., Mamatyuk V.I., Gatilov Yu.V., Bagryanskaya I.Yu. // Zhurn. organ.khimii. 1986. T. 22.Vyp. 6.S.1297-1306.
- Petrova T.D., Ryabichev A.G., Savchenko T.I., Kolesnikova I.V., Platonov V.E. // Zhurn. organ.khimii. 1988. T. 24.Vyp. 7. S.1513-1517.
- Petrova T.D., Platonov V.E.,Pokrovskij L.M. // Izv. AN. Ser. khim., 2002. N 3. S. 504-506.
- Petrova T.D., Platonov V.E., Maksimov A.M. // J. Fluorine Chem. 1999. V. 98. N. 1. P. 17-28.
- Kolesnikova I.V., Ryabichev A.G., Petrova T.D., Platonov V.E. //Izv. AN SSSR. Ser. khim. 1988. N 7. S. 1651-1654.
- Petrova T.D., Popova I.S., Kolesnikova I.V.,Platonov V.E. // Izv. AN SSSR. Ser. khim. 1994. N. 6. S. 1089-1094.
- Petrova T.D., Diss. dokt. khim. nauk. NIOKh SO RAN. Novosibirsk. 1995.
- Kolesnikova I.V., Petrova T.D., Platonov V.E., Ryabicheva T.G., Mihajlov V.A., Popov A.A., Savelova V.A. // Zhurn. organ. khimii.1989. T. 25. V. 8. S. 1689-1695.
- Kolesnikova I.V., Petrova T.D., Platonov V.E., Mikhailov V.A., Popov A.A., SavelovaV.A. // J. Fluorine Chem. 1988. V. 40. N 2-3. P. 217-246.
- Kryuchkova E.N., Kolesnikova I.V., Drizhd L.P., Savelova V.A., Petrova T.D., Platonov V.E. // Zhurn. organ. khimii. 1988. T. 24. V. 10. S. 2042-2046.
- Mihajlov V.A., Savelova V.A., Popov A.A., Petrova T.D., Platonov V.E. // Zhurn. organ. khimii. 2002. T. 39. V. 8. S. 1187-1195.
- Savelova V.A., Popov A.A., Petrova T.D., Platonov V.E., Mihajlov V.A. // Teoret. i eksp. khimiya. 2003. T. 39. N 1. S. 22-26.
- Petrova T.D., Platonov V.E. // Zhurn. organ. khimii. 1997. T. 33. Vyp. 5. S. 745-749.
- Petrova T.D., Kolesnikova I.V., Savchenko T.I., Platonov V.E. // Zhurn. organ. khimii. 1984. T. 20. V. 6. S. 1197-1204.
- Petrova T.D., Platonov V.E., Shchegoleva L.N., Maksimov A.M., Haas A., SchelvisM., Lieb M. // J. Fluorine Chem. 1996. V. 79. N. 1. P. 13-25.
- Petrova T.D., Kolesnikova I.V.,Mamatyuk V.I., Vetchinov V.P., Platonov V.E., Bagryanskaya I.Yu., Gatilov Yu.V. // Izv. AN. Ser. khim. 1993. N. 9. S. 1605-1611.
- Petrova T.D., Platonov V.E., Pokrovskii L.M., Rybalova T.V., Gatilov Yu.V. // Collect.Czech. Chem. Commun. 2002. V. 67. N. 10. P. 1449-1466.
- Petrova T.D., Platonov V.E., KolesnikovaI.V., Rybalova T.V., Bagryanskaya I.Yu.,Gatilov Yu.V. // J. Fluorine Chem. 2000. V. 103. N. 1. P. 63-73.
- Petrova T.D., Platonov V.E. // Zhurn. organ. khimii. 2005. T. 41. V. 2. S. 228-236.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V., Osina O.I., Gatilov Yu.V. // J. Fluorine Chem. 2001. V. 109. Is. 2. P. 131-139.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V, Osina O.I. // J. Fluorine Chem. 2002. V. 116. Is. 1. P. 3-8.
- Platonov V.E., Haas A., Schelvis M., Lieb M., Dvornikova K.V., Osina O.I., RybalovaT.V., Gatilov Yu.V. // J. Fluorine Chem. 2002.V. 114. Is. 1. P. 55-61.
- Platonov V.E., Bredikhin R.A., Maksimov A.M., Kireenkov V.V. // J. Fluorine Chem.2010.V. 131. Is. 1. P. 13-16.
Recommended for publication by Prof. V.E. Platonov
Fluorine Notes, 2011, 77, 1-2
