Received: June, 2010
Fluorine Notes, 2011, 76, 3-4
The method for the manufacture of 2,5-di(trifluoromethyl)-3,6-dioxa-8-(sulfonylfluoride)perfluorooctanoyl fluoride
N.N.Zatsepina, V.Z.Kaufman, A.E.Krivoshein, V.G.Barabanov, V.V. Kornilov
FCUE Russian Scientific Center "Applied Chemistry", 197198, Russia, St. Petersburg, Dobrolubov av. 14
e-mail: vbarabanov@rscac.spb.ru
Abstract: A method for the manufacture of 2,5-di(trifluoromethyl)-3,6-dioxa-8-(sulfonylfluoride)perfluorooctanoyl fluoride (FS-161) is developed that is based on the interaction between fluorosulfonyldifluoroacetyl fluoride perfluoroalkoxide and hexafluoropropyleneoxide in dry aprotic solvent using anhydrous potassium fluoride for catalyst. The factors that influence the selectivity of FS-161 formation in the process under study are investigated.
Keywords:oligomerization, sulfonyl fluoride, perfluorooctanoyl fluoride
2,5-Di(trifluoromethyl)-3,6-dioxa-8-(sulfonylfluoride)perfluorooctanoyl fluoride (FS-161) is a half-product in the synthesis of perfluoro(3,6-dioxa-4-methyl-7-octene)sulfonylfluoride (FC-141 monomer), and therefore it participates in the common process of Nafion type perfluoropolymer manufacture intended for the ion-exchange membranes [1]. The optimization of its synthesis conditions is a part of the general program aimed at streamlining of the process for perfluoropolymer manufacture.
Furthermore, thanks to high reactivity of fluoroanhydrides, easily convertible to some other functional groups through well-known chemical techniques, FS-161 can serve as raw material for the manufacture of various perfluoroderivatives with characteristics that suit their further application requirements (acids, salts, ethers, alcohols, nitriles, etc.).
FS-161 fluoroanhydride though described in patent literature [2-4] is poorly studied by now. It is producible through the interaction of Difluoro(fluorosulfonyl)acetyl fluoride (isomer of tetrafluoroethane-β-sultone) (FC-41) with two molecules of hexafluoropropylene oxide (HFPO) in dry aprotic solvent environment using some fluoride ion for catalyst [2, 3]. The reaction is an especial case of the common method for the manufacture of fluoroanhydrides, widely applicable in the synthesis of perfluorocarboxylic acid fluoroanhydrides of various structure [5-8] depending their specific features.
The objective of this study was to optimize the conditions for the conversion of initial reagents to the target product and to develop an efficient technological process for its synthesis.
The oligomerisation is described by the diagram as follows [5, 6]:
1. Stage of initiation
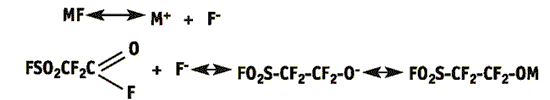
2. Stage of chain propagation and its limitation
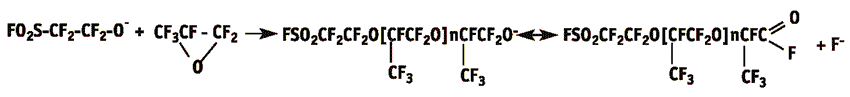
At the first stage fluoride-ion is attached to the carbon of FC-41 molecule carboxide and active perfluoroalkoxide-ion is formed that attacks electrophilic β-atom of carbon in HFPO at the next stage. This attack performs as the process factor that results in the ring opening and in the formation of hydroxyl-anion to which other HFPO molecules are added consequently resulting in oligomers. The chain termination depends on the mole ratio and other experiment conditions.
Oligomerisation is usually initiated by some alkali metal fluorides (KF, CsF). Tertiary ammonia fluorides, alkali metal halogenides or mixtures thereof (LiCl-CsF, LiCl-KF) are also applicable [2]. Some glymes (diglyme, triglyme, tetraglyme) or nitriles (acetonitrile, propionitrile) are frequently used for suitable polar aprotic solvents. It is essential that solvents are dried thoroughly and do not involve active proton-containing impurities. It is important that aprotic solvents are prone to specific salvation of cations so that anions remain free, thus providing the increase in the activity of fluoride- and perfluoroalkoxide anions in those reactions. HFPO oligomerisation exemplifies the obvious connection between the degree of oligomerisation and the solvent nature [6].
The process of FS-161 production in any case is followed by the formation of oligomeric by-products due to adding of one or three HFPO molecules to FC-41 (that results in FS-101 or FS-221, correspondingly). The possibility of parallel isomerisation or oligomerisation of HFPO must be taken into account as well. The mechanism of fluoroanhydride oligomerisation is similar to that of HFPO, and the predominant direction of those reactions depends on the type of the reagents that are mostly attacked by fluoride-ions. As a rule, the reactivity of fluoroanhydrides exceeds that of HFPO significantly. Special conditions must be chosen so that to limit the formation of by-products (FS-161 and HFPO oligomers) and to favour the increase in the yield of FS-161 fluoroanhydride.
We studied the impact of various factors on the yield of the target product with the aim to optimize those conditions. Possible variation in the reaction conditions involved the choice of catalysts, solvents, mole ratio of the reagents, temperature regime, pressure, etc. From the typical tests results shown in the table below some conclusions can be reached about the influence of the said factors on the process under study. The chromatographic analysis of the fluorinated layer resulting from the oligomerisation serves for the quantification of relative content of FS-161 fluoroanhydride and impurities.
1. FC-41 to HFPO mole ratio belongs to the factors governing the molecular–mass distribution of oligomers within the product layer. Its variation allows obtaining of either the target fluoroanhydride (FS-161) or oligomers (FS-101 and FS-221) for the main products. However, one always has to deal with some oligomer mixture, and its composition depends largely on the experiment conditions. Usually, the final product contains 5-10% FS-101 and 10-20% FS-221 (see Table). FS-101 is a half-product in FS-161 synthesis and undergoes recycling (exp.10, 11). The share of FS-221 oligomer impurity grows if HFPO feeding is too rapid, mixing inefficient (exp.9), and temperature regime is violated. Decreasing the mole ratio FC-41: HFPO to 1.8-1.9 (instead of stoichiometric relationship 1:2) it is possible to reduce partly the formation of "heavy" FS-221 oligomer.
2. From the Table below one may see that both tested catalysts (KF, CsF) display high and nearly similar catalytic activity (compare exp.5 and exp.6). However, potassium fluoride is preferable as more easily available and less expensive. The best mole ratio is FC-41 : KF : HFPO = 1 : 0.1-0.15 : 1.8-1.9. This amount of the catalyst unchanged during the reaction is usually sufficient for efficient oligomerisation. Its larger quantity besides of possible impact on the process selectivity may cause the loss in the total mass of the product layer (due to passing of additional amount of perfluoroalcoholates into aprotic solvent).
3. The optimal temperature range for the oligomerisation of FC-41 alkoxide is 15-25ºC. At lower temperatures (≤ 0ºC) there is obvious uptrend in the share of "heavy" (FS-221) oligomer (exp.7) in the product layer; at higher temperatures there is a notable impact of the concurrent by-process that is the formation of low-molecular HFPO oligomers (dimers, trimers) (exp. 8).
The observed picture represents the influence of the temperature regime on the catalyst system properties. It is well-known that the stability of alkali metal perfluoroalkoxides decreases with increasing temperature [5]. It is conceivable that deviations from the optimal temperature range may cause either increase in the rate of propagation at low temperatures, or its limitation and low-molecular products formation at higher temperatures.
4. We studied the medium impact on the selectivity of FS-161 formation was using three polar aprotic solvents (diglyme, tetraglyme, acetonitrile) for examples. Just those solvents are the most often recommended for oligomerisation. As the results are compared (exp. 10-16) one may see very strong differentiating influence of the solvents on the product oligomer layer composition. The behaviour of glymes (diglyme, tetraglyme) is nearly the same; they favour predominantly the target product formation. Tetraglyme is more efficient (higher selectivity, softer oligomerisation conditions); however, diglyme is preferable in practice being more easily available. In the presence of acetonitrile the yield of FS-161 drops dramatically due to the significant amounts of "light" oligomers in the product layer (FS-101, dimers and trimers of HFPO, etc.); this solvent, consequently is non-applicable in the process under study. One possible explanation of the said different solvent effect is the difference in their solvation characteristics. It is possible that rather small to compare with glymes specific salvation of acetonitrile causes the destabilization of perfluoroalkoxide-anion thus favouring both the chain termination and "light"” oligomers formation.
The quantity of solvent, apparently, is not a limiting factor. It must be sufficient for at least partial solution of potassium fluoride, HFPO and its perfluoroalcoholates and for intensive stirring of the reagents during the oligomerisation process (the best mole ratio FC-141: diglyme = 1 : 0.5-0.7).
Such composition saturated with perfluoroalcoholate oligomers may be used for a catalyst in 3-4 consecutive tests. As the catalyst activity drops the product should undergo fractional distillation. The regenerated oligomer mixture is added to the product layer, and solvent (diglyme) is recycled.
5. From the Table data one may see that FS-161 fluoroanhydride manufacture process is successfully carried out either in a glass reactor under atmospheric pressure, or in an autoclave under small surplus pressure of HFPO (0.3-0.5 atm). When so doing, the pressure value is not critical.
Table. The impact of reaction conditions on the selectivity of FS-161 formation.
|
Experiment # |
Mole ratio |
Reaction conditions |
Content of main products in product layer, % (GLC)2) |
|||||
|
FC-41:KF: HFPO: DG1) |
T, ºC |
P, atm |
τ, hour |
DG1) |
FS-101 |
FS-161 |
FS-221 |
|
|
1 |
1:0,12:2:0,7 |
15-20 |
- |
1,5 |
1,5 |
5 |
74 |
19 |
|
23) |
1:0,12:1,9:1 |
15-20 |
- |
3,5 |
0,6 |
6 |
78 |
14 |
|
33) |
1:0,15:1,8:0,6 |
18-22 |
- |
2 |
1 |
8 |
78 |
13 |
|
43) |
1:0,1:1,85:0,7 |
18-22 |
- |
3 |
- |
11 |
80 |
9 |
|
5 |
1:0,12:1,8:1 |
5-10 |
- |
2,5 |
- |
8 |
76 |
15 |
|
6 |
1:0,12:1,8:1(CsF) |
5-10 |
- |
1,5 |
- |
9 |
74 |
16 |
|
7 |
1:0,15:1,8:1 |
-5-5 |
- |
2 |
- |
9 |
69 |
21 |
|
8 |
1:0,1:1,8:1 |
25-40 |
- |
1,5 |
8 |
12 |
67 |
12 |
|
94) |
1:0,12:1,8:1 |
15-22 |
- |
2 |
- |
6 |
67 |
26 |
|
10 |
1:0,15:1:1 (FS-101) |
15-20 |
- |
1,5 |
- |
8 |
79 |
13 |
|
11 |
1:0,1:1:0,6(FS-101) |
18-25 |
- |
2 |
- |
10 |
81 |
9 |
|
12 |
1:0,13:1,9:3,5(CH3CN) |
18-23 |
- |
3 |
16 |
21 |
53 |
3 |
|
13 |
1:0,13:1,9:20 (CH3CN) |
18-23 |
1,5-0,3 |
4 |
21 |
32 |
43 |
4 |
|
14 |
1:0,13:1,9:0,5(TG1)) |
18-23 |
0,3-0 |
2 |
1 |
8 |
80 |
10 |
|
153) |
1:0,11:1,9:0,6 |
15-18 |
0,5-0,1 |
3 |
2 |
6 |
77 |
15 |
|
163) |
1:0,15:1,9:0,6 |
18-23 |
0,5-0 |
2,5 |
- |
6 |
78 |
16 |
Notes: 1) DG is diglyme, DM is HFPO dimer, TG is tetraglyme. 2) Conversion of original reagents is 98-100%. 3) Optimized conditions. 4) Inefficient stirring by magnetic stirrer.
The study results were used for the optimization of the conditions of the FS-161 fluoroanhydride production process developed in laboratory scale. The process regime is described in the Experiment chapter. The technology enables the manufacture of FS-161 with the yield of 70-75% and purity at least 99% (by GLC).
Experiment
Original reagents
The success of the synthesis depends mostly on the cleanness and drying of original reagents. To remove the crystal water potassium fluoride (pure) was heated to 140-160ºC, abraded carefully and calcined additionally during 4-6 hours under vacuum at 250-300ºC.
Solvents (diglyme (99%), tetraglyme (98%), and acetonitrile (99%)) were dried and kept above molecular sieves 4А (water content ≤ 0.05% according to GLC).
FC-41 (≥98%) (produced by Moldavsky et al. [10]) and HFPO (≥98.5%) were used without further purification. All apparatus was dried carefully and flushed with dry inert gas (nitrogen, argon).
Manufacture of FS-161
The process was conducted in areactor of volume 1 dm3, equipped with a stainless steel jacket and a driver with a stirrer (up to 750 rpm), vacuum-meter, pocket for thermocouple, reagent feeding valves and a bottom bleeding valve.
Anhydrous potassium fluoride (12 g) and absolute diglyme (150g) were charged through an access hole into the apparatus preliminary flushed with inert gas. The resulting mixture was stirred during 1 hour. Then the reactor was evacuated at minus 5-0ºC, and 300g of FC-41 were added during 1 hour through an addition funnel connected to its valve. To complete the formation of FC-41 alkoxide the reaction mixture was further stirred during 1 hour at 10-15ºC. After that HFPO (520g) was fed during 2.5-3.5 hours under intensive stirring at 15-25ºC through a siphon valve from a cylinder placed on scales; the feeding rate was chosen so that the apparatus pressure would be 0.3-0.5 atm and decreased gradually as HFPO spent. After feeding of the calculated quantity of HFPO the resulting mixture was further stirred during 1-2 hours till its pressure is stable, and kept for one night. Next day the reactor was cooled during 1-2 hours at minus 10 - minus 20ºC without stirring in order to improve the product stratification into two layers: the bottom fluoroanhydride layer (75%-80% FS-161) and the top diglyme-alcoholate layer.
Through the bottom bleeding valve the product layer was discharged into a collector for further rectification. The remaining diglyme-alcoholate layer was recycled twice or three times to the FS-161 synthesis stage. Then after fraction distillation cleaning under vacuum diglyme was returned into the technological cycle; the mixture of fluoroanhydrides separated from solvent stripper was added to the product layer. FS-161 fluoroanhydride was separated by fraction distillation (vapour temperature 82-84ºC at 75 mm Hg column; boiling temperature 148-151ºC). Intermediate factions containing FS-101 and non-conditional FS-161 were returned into the technological cycle. The still residue contained FS-221 and other heavy impurities.
The technological yield of FS-161 fluoroanhydride (taking into account recycling of intermediates) was 540-580g (70-75% of theoretical value); part by mass of the main substance was at least 99% (according to GLC). Both IR and 19F NMR spectra confirmed that the product corresponded in structure to the formula of FS-161.
Its IR spectra included bands (ν, cm-1):
1864(COF), 1468 (SO2Fas),
region 1050-1350 (CF2,CF3 – groups).
19F NMR spectrum of F9SO2CF28CF27OCF6(CF35)CF24OCF3(CF32)CF1O
(δ, ppm; yF-F, Hz):
- 26,1 (t, 1F-2F 13,0); 2) -81,3 (d2, 2F–1F 13,0); 3) -130,2 (t, 3F–4F 20,4); 4)-80,6 (m, 5F–4F 7,6; 4F–3F 20,4); 5) -82,3 (d, 5F–4F 7,6); 6) -145,6 ( t, 6F–7F 19,3); 7) -92,0 (m, 7F-9F 13,4; 7F–6F 19,3); 8) -115,9 (t, 8F-7F 4,0); 9) 42,0 (t, 9F–7F 13,4)
Following the same method FS-161 was produced at atmospheric pressure. The process was conducted in a glass 5-necked flask equipped with a dropping funnel, siphon for HFPO feeding, thermometer, reflux condenser cooled with dry ice, and stirrer with cylindrical grinded neck providing both intensive stirring and apparatus sealing.
Conclusions:
A method is developed for the manufacture of fluoroanhydride 2,5-di(trifluoromethyl)-3,6-dioxa-8-sulfonylfluorideperfluorooctanoyl fluoride with the main substance content ≥99%, intended for the synthesis of FCS-141 monomer.
References
- L. F. Sokolov, A.S. Odinokov, O.S. Bazanova in: V.V. Kornilov, B.N. Maximov (Eds.) "Fluorine compounds. Chemistry, Technology, application", Teza, Saint-Petersburg, 2009, p. 109-111
- US Patent 3301893, 1967
- US Patent 5463005, 1995
- Patent Russian Federation 2269513, 2006
- V.A.Ponomarenko, S.P.Krukovsky, Yu.A.Alybina. Fluorine containing heterochain polymers, Nauka, Moscow, 1973, p.56-80
- H. Millauer, W. Schwertfeger, S. Siegemund. Angewandte Chemie. 1985, Vol.97,N 3, p. 164-182
- G.G. Jakobson, V. V. Bardin, Fluoride-Ion in organic chemistry, Nauka, Novosibirsk, 1986, p. 287-293
- I.L. Knunyants and G.G. Jakobson, Editors, Synthesis of fluoroorganic compounds, Khimiya, Moscow (1977) p. 128-136
- A.A. Glazkov, A.V. Ignatenko, S.P. Krukovsky, V.A. Ponomarenko, Izv Akad Nauk SSSR Khim, 1979, V. 11, p 2512-2515
- V.G. Barabanov, T.A. Bispen, A.N. Ilyin et al, The Improved Preparation Method of Difluoro(fluorosulfonyl)acetyl fluoride, Fluorine notes, Vol. 2(75) 2011, /public/2011/2_2011/letters/letter4.html
Recommended for publication by V. Kornilov
Fluorine Notes, 2011, 76, 3-4
