Fluorine Notes, 2011, 74, 1-2
DEHYDRATION OF METHANOL ON THE CATALYST BASED ON PERFLUORINATED COPOLYMER F-4SF
N.V.Kolesnichenko, A.B.Kulikov, A.L.Maximov, A.I.Nehaev, R.V.Kulumbekov , V.G.Barabanov*, V.V.Kornilov*
A.V.Topchiev Institute of Petrochemical Synthesis, RAS, 119991, Russia, Moscow, Leninsky prospect 29,
e-mail: kipnis@ips.ac.ru
*FSUE Russian Scientific Center
"Applied Chemistry", 197198, Russia, St. Petersburg, Dobrolubov av. 14
Abstract: A series of acid catalysts comprising perfluorinated copolymer F-4SF on various supports are synthesized. The catalyst was characterized by transmission electron microscopy, infrared spectroscopy and thermogravimetry; the characteristics of its porous structure are shown. The possibility of its use for a heterogeneous catalyst in the conversion of methanol to dimethyl ether (DME) is demonstrated. It is found that at 190 °C in a flow mode, the catalyst retained its activity for 10 h, and the methanol conversion was 90% with 100% selectivity by DME
Keywords:perfluorinated copolymer, dimethyl ether, catalysts
Introduction
Not so long ago the major interest to DME was connected with its use in aerosol production, mainly for cosmetic applications. Recently it attracted more attention as a possible substitute for diesel fuel [1]. It is particularly relevant in South-East Asia, where hydrocarbon feedstock is not available, and coal or imported liquid natural gas should be used for raw materials in chemical industry. The benefits of DME are is that it is non-toxic and does not cause corrosion, and burns smokeless without emission of sulfur oxides or nitrogen. DME may be used for feedstock in the lower olefins manufacture with zeolite-containing catalysts: propylene and ethylene that are important half-products in chemical industry.
One of the methods for DME manufacture is the dehydration of methanol:
2CH3OH ↔CH3OCH3 + H2O
This catalitic process follows an acid mechanism. γ-Al2O3, modified HY and H-ZSM-5 zeolites [2], aluminophosphates [3], etc. were studied for the reaction catalysts.
In [4] the authors proposed to use thermostable sulfoionite catalysts in the dehydration of methanol to pure DME at 110-150oC and pressure 4,9-13,2 atm in a combined reaction-rectification apparatus, involving two rectification zones and interim reaction zone filled with the catalyst. Other researchers [5, 6] studied the dehydration of methanol on perfluorinated ion-exchange polymer Nafion (developed by DuPont) tested earlier with acid catalysts [7, 8].
F-4SF is a Russian fluoroplast product, analogue of Nafion that is a perfluorinated sulfogroup-containing co-polymer of 2 monomers: tetrafluoroethylene and perfluoro-3,6-dioxy-4-methyl-7-octene-sulfoacids. F-4SF polymer is not water-soluble, non-toxic, thermostable up to 300oC, and only its sulfogroup is more or less reactive (hydrolysis results in the conversion of its sulfonylfluoride groups –SO2F to sulfoacid groups –SO3H).
The acidic form of F-4SF co-polymer is a strong solid acid, and its properties are due to the effect of electron transfer from hydrophobic perfluorocarbon groups to hydrophilic sulfoacid groups. Its Hammett acidity is –12, that is comparable to 100% sulfuric acid. [9]. However, this co-polymer does not cause corrosion of equipment as its acid sulfogroups are firmly bonded to the polymer. The specific surface area of F-4SF is rather small (less than 0,2 m2/g), and it puts upper limit to the co-polymer catalytic activity. To increase the surface area one may deposit F-4SF on a porous support with developed surface, e.g. during the synthesis as it was done earlier in the case of Nafion-based catalysts manufacture [5-8,10-12]. Those Nafion-silicon dioxide-composites had significant surface area (150-800 m2/g) and involved nano-sized (< 100 nm) polymer particles incorporated into the carcass of silicon dioxide pores.
In [13], e.g., a catalyst was prepared with Nafion inserted into mesoporous silicate MCM-41 in the process of its synthesis using Nafion 5% solution in ethanol, tetramethylammonia hydroxide, tetraethoxysilane for the source of silicon and cetyltrimethylammonia bromide for a template. After blending of those components the reaction mixture was autoclaved at 130°C, the residuum was then separated from spent liquor and washed with water. The template was removed by treatment with concentrated sulfuric acid and then washed with ethanol. The resulting catalyst was dried at 150°C. A group of researchers [6] synthesized Nafion/MCM-41 composite with surface area 800 m2/g. In patent [14] a catalyst composition based on Nafion and silicon oxide with specific surface area 200m2/g was produced by a sol-gel method from tetramethoxysilane 5% solution of Nafion in lower alcohols.
Nafion-based catalysts were used in the dehydration of methanol. Therefore, in [5] the conversion of helium-diluted methanol achieved 40% with quantitative selectivity by DME at 220°C.
The objective of this study was to develop an approach to the synthesis of catalysts for DME manufacture via the heterogenezation of F-4SF polymer on various supports.
Experimental
The samples of catalystcontainingthedesigned amount ofsupported F-4SF perfluorosulfopolymer were produced according to the technique as follows.
Under intensive stirring 93 ml of 0,4 mol/l NaOH solution and 75 ml of water were added to the designed solution of perfluorosulfopolymer (containing 7,2% of F-4SF with equivalent mass (EM) 890 in iso-propanol). The resulting solution at room temperature was distilled under vacuum with the help of a rotor evaporator to the volume of 330 ml.
In another flask 20 ml of water and two drops of concentrated hydrochloric acid were added to 138 g of tetraethylortosilicate. The mixture was stirred for 15 minutes at room temperature to obtain a uniform colloid solution, with simultaneous massive gas emission. As the solution temperature rose significantly we cooled it to room temperature with cold water. After cooling to room temperature the solution under intensive stirring was poured into the perfluorosulfopolymer solution by thin jet.
The solution was stirred intensively at weak heating and the formation of gelatinous deposit was observed
in a few minutes.
This gel was kept in air at room temperature for 8 hours, and dried at
95°C during 2 days to moisture-airfree condition. The resulting composite was treated with 300 ml
of 15% nitric acid at 70°C during 7 hours. Solids were then separated from solution by filtration,
washed with distilled water and dried under vacuum at 100°C during 24 hours to receive solid glassy
material with mass 59 g (95% yield).
To determine the amount of acidic sites in the catalyst we used the method of acid-base titration. An assay of the sample was being stirred intensively during 4 hours in 10% water solution of NaCl, then its solid phase was separated by filtration, and the solution was titrated with 0,01000 mole/l NaOH solution (phenolphthalein was used for the indicator).
The characteristics of the sample porosity were detected with the help of ASAP – 2010N (Micrometrics) analyzer. Prior to analysis the sample was evacuated at 200°C during 6 hours to pressure 1*10–3 atm. The nitrogen adsorption-desorption isotherm was registered at 77 The porosity characteristics were calculated with the help of ASAP – 2010N standard software. For the samples characteristics we used the surface areas (BET method), volumes (at ð/ð0=0,95) and the pore diameters. In order to obtain the pore distribution curve we used the BJH method.
IR-spectra were recorded using IFS-60 v/s Bruker Fourier spectrometer (scan number was 50). OPUS and ORIGIN software was used in spectra processing. The sample was pelletized with KBr.
Thermal stability of the sample and its composition were studied by thermal gravimetry in air at dynamic heating regime in Q-1500 D derivatograph (Hungary). The sample mass was 100 mg, aluminum oxide was used for standard. The heating rate was 10°C/min, the crucible was made of platinum.
Thus produced samples were studied by the method of X-ray photoelectron spectroscopy (RPHES) using LAS – 3000 electronic instrument equipped with a photoelectron analyzer with OPX-150 retarding potential. The photoelectron excitation was carried out using X-ray emission of an aluminum anode (Al Kα = 1486,6 eV) with tube voltage 12 kV and emission current 20 mA. Photoelectron peaks were calibrated by carbon C 1s line with binding energy 285 eV.
The studies by the methods of transmission electron microscopy (TEM) and characteristic electron energy loss spectroscopy (CEELS) were conducted with the help of LEO912 AB OMEGA transmission electron microscope.
Catalytic tests in the synthesis of dimethyl ether from methanol were done in a flow-through catalytic unit at atmospheric pressure. Prior to the reaction the catalyst was fractioned; its fraction with particle size more than 1 mm was separated and dried in vacuum at 100°C. The bulk rate of methanol flow was 1,0 h–1.
The catalyst was activated directly in the reactor. To this end it was heated in nitrogen flow to 200°C, and kept at this temperature for 2 hours to remove humidity traces, then the reactor temperature was decreased to required value and the catalyst testing started.
Chromatographic gas analysis was conducted with Porapak T column (Crystallux 4000 chromatograph, catharometer). The sampling was done hourly upstream to the reactor at temperature no less than that of the reactor (before water and methanol condensation in traps). The possible presence of Î2, H2, CO, and CO2 impurities was controlled by the gas flow condensation at 0°C in columns packed with molecular seives (13X and Polysorb) of CHROM 5 chromatograph. The samples for analysis were taken once every hour.
Relying on the chromatography results we calculated the methanol conversion as follows:
K = ((mCH3OHinlet – mCH3OHexit)/mCH3OHinlet)*·100%,
here K – methanol
conversion, %;
mCH3OHinlet – mass of methanol fed into the reactor, g;
mCH3OHexit – mass of methanol at the reactor exit, g.
DME yield was calculated as follows:
B =K*mCH3OH->DME/mCH3OH,
here B
– DME yield, %;
mCH3OH->DME – mass of methanol converted to DME, g;
mCH3OH – mass of reacted methanol, g.
Results and discussion
Synthesis and physico-chemical properties of catalysts
The catalysts were synthesized through the insertion of the polymer into the support matrix during its in-solution formation by the sol-gel method. Application of the said method provides location of the polymer particles in the forming support "network" with pores much less in size than polymer molecules or aggregates. It results in large general surface area of the matter and the area of contact between the polymer active sites and reagents grows due to peculiar "spraying" of polymer in the matrix during its synthesis.
The catalyst formation is based on the hydrolysis silicon-containing substance in aqueous or aqueous-alcohol solution of F-4SF perfluorinated polymer resulting in the formation of silica gel. Here the gelation follows the mechanism of tetraethoxysilane polycondensation in the process of its hydrolysis:
Si(OEt)4 + 4nH2O → nSi(OH)4 + 4nEtOH
2nSi(OH)4 → nSi(OH)4—SinO2n-m + (2n-m)H2O
Primary sol particles sized approximately 2-20nm in the process of their drying form a network structure of linked spherical particles. As the number of particles grow and their links strengthen a robust silicon-oxygen carcass if formed. During the gelation both micelles and polymer aggregates present in the solution are fixed between the said particles and in the process of drying they are brought into the carcass cavities. Those pores being the intervals between the particles their size depends on the size of silica gel particles. This size being significantly less than even that of the polymer monomolecular micelles it is easily fixed in the material.
In the interim solid product the polymer has salt (sodium) form. In order to convert it to acidic form and to remove possible contaminations, and residual organic substances, e.g., solvents, it must be treated with nitric acid solution as acidic catalyst does not exhibit activity in their salt form:
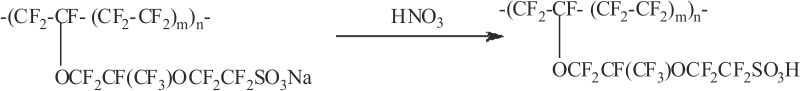
We prepared a number of catalyst samples that differ in the content of polymer (13%, 25%, 30%, and 40%) and in their bulk capacity. Physico-chemical properties of the produced samples are shown in Table 1.
Table 1. Content of polymer in samples and their bulk capacities
| Sample |
Theor.cont., % | Content by TGA, % | Measured F/Si ratio by RPHES |
SBET |
Average pore size, Å |
Pore volume, cm3/g | Exchange capacity, meq/g |
| 13% Ô-4ÑÔ/SiO2 |
13 |
14 |
0,58 |
395 |
61 |
0,81 |
0,14 |
|
25% Ô-4ÑÔ/SiO2 |
25 |
26 |
1,3 |
197 |
126 |
0,80 |
0,27 |
|
30% Ô-4ÑÔ/SiO2 |
30 |
31 |
1,8 |
241 |
61 |
0,41 |
0,32 |
|
40% Ô-4ÑÔ |
40 |
41,5 |
2,6 |
231 |
131 |
0,73 |
0,42 |
The intimacy of polymer involving into the synthesis was determined by the results of thermogravimetric analysis (Fig.1).
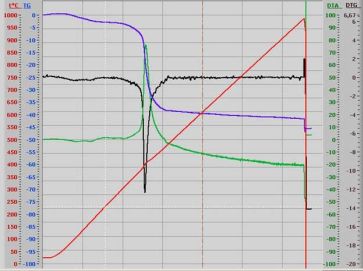
Fig.1. Thermogram of the catalyst sample with 40% of polymer
Water that still remained on the sample surface even after evacuation at 100°C is removed when the composite is further heated up to 200°C. This water makes about 2,5% of the sample mass. At temperature above 250°C the decomposition of perfluorinated co-polymer from composite begins and the total mass loss at this second step corresponds to its content (Table 1).
The presence of polymer in our material was confirmed by RPHES spectra as well. Three fluorine signals (685.2, 686,7, 688,6 EV) and one for carbon (284,9; 286,5; 288,2 EV) are typical for perfluorinated F-4SF polymer. The fluorine to silicon ratio of atomic surface concentrations makes it possible to estimate the concentration of polymer on the surface, and it was close to the calculated value.
In all samples the amount of polymer was also detected titrometrically by the number of involved acid groups and using their RPHES data (table 1). It should be noted that the results coincided accurately with the calculated values for the tetraethoxysilane-based catalysts. Thermogravimetric analysis allows the most accurate determination of the polymer content by the mass loss during its decomposition. RPHES makes it possible to assess the polymer content from the intensity ratio for peaks that correspond to fluorine and silicon binding energies. The exchange capacities for the catalyst H+ ions for the catalysts correspond to equivalent mass of the polymer in solution (890).
According to the titration results the catalyst bulk capacities were proportional to the polymer content and were 0,14-0,42 meq/g. The data on bulk capacities of H+ ions for catalysts with equivalent polymer mass in solution correspond to nearly complete involvement of the polymer into the composite taking into account its amount introduced during the synthesis. Those data evidence that acidic sited of the material are easily accessible.
The results of our study of the catalyst sample using the method of transmission electron microscopy are shown in the Fig.2. Those data confirm both the homogeneity of those samples, and uniformity of the polymer distribution over the support surface for all samples with various content of the polymer. In the case of tetraethoxysilane a dendrite structure is formed based on small (2-5 nm) particles of silicon oxide. Accordingly the sizes of polymer particles and aggregates included into the material structure are also small.
 |
|
|
13% F-4SF supported |
40% F-4SF supported |
Fig.2. TEM-data for the catalyst samples: 40% F-4SF and 13% F-4SF
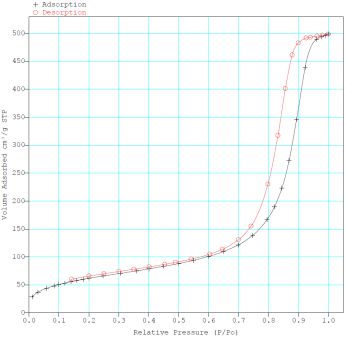
The results of the surface area and porosity measurements are exemplified by F-4SF 40% polymer (Figure 3).
 |
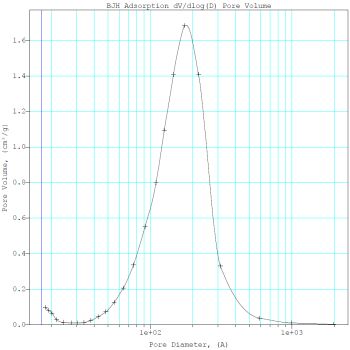 |
Fig.3. Nitrogen adsorption-desorption (a) and pore size-distribution (b) in a sample of catalyst with 40% polymer content
The results of the surface area and porosity measurements are exemplified by F-4SF 40% polymer (Figure
3).
The catalyst surface area was between 197 and 351 m2/g, the average pore diameter
being 61-131Å. Therefore it may be classified with mesoporous substances. Pore size distribution
is rather wide, and besides of mesopores a small number of micropores and macropores are also present.
The pore volume in the produced materials varied between 0.41 to 0,73 cm3/g.
The
IR spectroscopy results are shown in Figure 4.
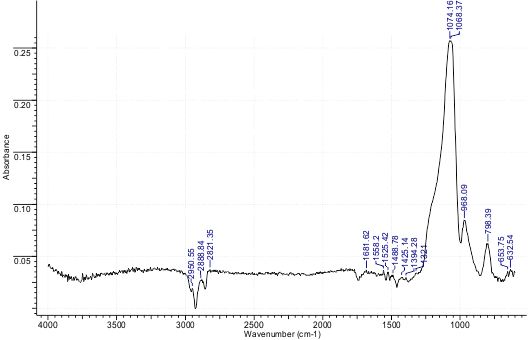
Fig.4. Example of IR spectrum for a sample of the catalyst (40% polymer)
The presence of the polymer in samples is revealed by the presence of spectral bands peaked at 1161 and 1123 cm–1, while the spectrum of pure silicon oxide involves a band at 1098 cm–1. The most intensive IR spectral bands of the sample shown in Fig.5 are located within the interval 1065-1075 cm–1. Here we have strong superposition of a number of bands that belong to Si-O-Si (SiO2), C-F and S=O bonds of F-4SF polymer. The band at 1394 cm–1 assigned to nitrate ion is very weak thus pointing to only traces of NO3-. Low-intensive bands at 600-700 cm–1 indicate that the polymer in the catalyst is in its acidic form.
Testing of the catalysts in the process of methanol conversion
Primary data of the study on the catalyst samples demonstrated that within the temperature interval 70-120°C high initial conversion of the substrate has place, but the reaction is followed by rapid deactivation of the catalyst. Our further studies have shown that the sample deactivation is related to tight sorption of methanol. Further experiments lead to the conclusion about the most strong sorption of methanol on the polymer surface, partly, possibly, with water; water is less strongly attached to the polymer surface, and DME is even less strongly than water. Therefore, to achieve considerable methanol conversion and to prevent deactivation the conditions are required that would provide growth of the rate of alcohol and water desorption, this being first of all higher reaction temperature. Further experimental studies on the catalytic activity of F-4SF-based composite were conducted within the temperature interval 130-190°C.
Those F-4SF-based samples were tested in the reaction of methanol dehydration and in a flow-through reactor for the development of Initial Data. In all cases the selectivity by DME was 100%. The data on substrate conversion at two chosen temperatures for the catalysts with variable polymer contents are shown in Fig.5. The conversion values at different temperatures on the catalyst with 25% or 40% polymer or pure polymer in acidic form are presented in table 2.
a)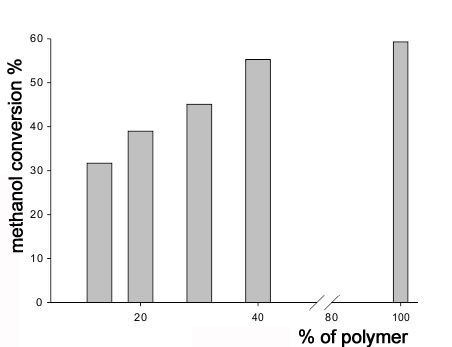
b)
Fig. 5. Dehydration of methanol resulting in dimethyl ether formation. WMEOH = 1hour-1, P = 0.1 MPa , a) 150 °C; b) 190°C
The conversion value grows with temperature and exceeds 90% at 190°C. It should be mentioned that the difference in the activity for catalysts with different content of polymer as to compare with pure polymer is high at 150°C but goes down as temperature grows. Apparently, for those hybrid materials the impact of diffusion limitations decreases and water desorption facilitates (as to compare with pure F-4SF catalyst) as temperature grows. It results in considerable increase in the specific catalytic activity of composite catalyst taking into account the number of active sites per 1 gram of the material. The difference in activity between F-4SF and the catalyst with 40% F-4SF/SiO2 is quite small.
Table 2. Conversion of methanol at various temperatures
| Catalyst |
Conversion, % |
|||
|
130°C |
150°C |
170°C |
190°C |
|
|
F-4SF |
20,8 |
59,3 |
68,6 |
92,2 |
|
25% F-4SF/SiO2 |
14,7 |
38,8 |
58,2 |
86,7 |
|
40% F-4SF/SiO2 |
20,1 |
55,3 |
68 |
90,2 |
From the above table 2 one may see that the maximal conversion of methanol on the composite material is 90,2% and observed at 190°C. The time dependence of the catalyst stability was tested at this temperature. The test results for methanol dehydration F-4SF-based composite catalysts at 190°C are shown in Figure 6.

Fig.6. Test results for a methanol dehydration F-4SF-based composite catalyst.
For the dehydration of methanol at 190°C the conversion was about 90%, and the selectivity by dimethyl ether was 100%. The process characteristics remained unchanged during all test period. The catalyst activity remained unchanged during its reuse after cooling. It must be noted that the catalysts now applied in the methanol dehydration usually work at higher temperatures.
Thus, under the conditions similar to those of Table 1 (130–190°C) the conversion of methanol on a sample of γ-Al2O3 (Production Association "Azot", Dneprodzerjinsk) and the catalyst activity was practically zero.
It also should be underlined that with F-4SF we succeeded to produce much more active catalysts than the Nafion-based composites. The conversion of methanol on Nafion/MCM-41 catalyst [6] reached its maximum (40%) at 300°C, while in [5] the same 40% were achieved at much lower (220°C) temperature.
Conclusions
It is shown that the proposed method for the composite synthesis makes it possible to produce an efficient catalyst for the dehydration of methanol. The tests resulted in a conclusion that the catalyst containing F-4SF perfluorinated polymer functioned for 10 hours without loss in activity, and made it possible to achieve 90% conversion of methanol with 100% selectivity by dimethyl ether. The proposed catalyst is more active in the dehydration of methanol to dimethyl ether than other catalysts currently in use, and it is applicable at lower temperatures.
References
2. Fu Y., Hong T., Chen J., Auroux A., Shen, J. Surface acidity and the dehydration of methanol to dimethyl ether. // Thermochim. Acta. 2005. V.434. N.1-2. P.22-26.
3. F. Yaripour, M. Mollavali, Sh. Mohammadi Jam, H. Atashi. Catalytic dehydration of methanol to dimethyl ether catalyzed by aluminum phosphate catalysts // Energy Fuels, 2009. V.23. N.4. P.1896–1900.
4. Patent RU 2282613, 2006.
5. Varisli D., Dogu T. Production of clean transportation fuel dimethylether by dehydration of methanol over nafion catalyst. // G.U. Journal of Science, 2008. V.21. N.2. P.37-41.
6. Ciftci A., Sezgi N.A., Dogu T. Nafion-incorporated silicate structured nanocomposite mesoporous catalysts for dimethyl ether synthesis // Ind. Eng. Chem. Res. Publication Date (Web): January 15, 2010.
7. Harmer M.A., Farneth W.E., Sun Q. High surface area Nafion resin/silica nanocomposites: a new class of solid acid catalyst // J. Am. Chem. Soc. 1996. V.118. N.33. P.7708-7715.
8. Wang H., Xu B.Q. Catalytic performance of Nafion/SiO2 nanocomposites for the synthesis of r-tocopherol // Appl. Catal., A. 2004. V.275. P.247-255.
9. Encyclopedia of Chemical Processing. NY: Taylor and Francis E.D. by S.Lee.
10. Ledneczki I., Darányi M., Fülöp F., Molnár A. SAC-13 silica nanocomposite solid acid catalyst in organic synthesis // Catal. Today. 2005. V.100. N.3-4. P.437-440.
11. A. Heidekum, M.A. Harmer, W.F. Hoelderich. //J.Catal. 1998. V.178. P.260.
12. M.A. Harmer, W.E.Farmeth, Q.J.Sun. //Adv. Mater. 1998. N.10. P.1225.
13. Fujiwara M., Kuraoka K., Yazawa T., Xu Q., Tanaka M., Souma Y. Preparation of an MCM-41/Nafion Composite Material; A selective catalyst for r-methylstyrene dimerization // Chem. Commun. 2000. N.16. P.1523-1524.
14. WO 1995/19222, М.А. Harmer - 1995
Recommended for publication by Ph.D. Vadim V. Kornilov
Fluorine Notes, 2011, 74, 1-2
