Fluorine Notes, 2010, 72, 1-2
Fluorine notes, Номер 5(72) 2010
Особенности прямого фторирования тетрафторэтилена в твердых и жидких перфторорганических средах
С.Р. Аллаяров,1 И.П. Ким,1 И.В. Маркин,2 А.А. Карнаух,1 И.М. Баркалов ,1 Д.А. Диксон.3
1) Институт Проблем Химической физики РАН, г.Черноголовка, Россия
2) "ФГУП
Российский научный центр "Прикладная Химия", пермский филиал", г. Пермь, Россия
3) Химический Факультет Алабамского Университета, г. Таскалууса, США
АННОТАЦИЯ.
Рассмотрена возможность организации безопасного фторирования тетрафторэтилена
(ТФЭ) в твердых и жидких перфторорганических средах. Процессы наблюдались методами низкотемпературной
калориметрии и ЭПР-спектроскопии. Состав рабочих смесей анализировался до и после экспериментов.
Фторирование кристаллических образцов ТФЭ и гексафторпропилена (ГФП) в отсутствии разбавителя
протекает в жестком взрывном режиме. В качестве разбавителей фторирования ТФЭ в твердофазных смесях
использовали олигомеры ГФП – димер (ГФП)2 и тример (ГФП)3.
Показано, что
жесткость режима падает в ряду систем: ТФЭ, [ТФЭ-(ГФП)2], [ТФЭ-(ГФП)3]. В последнем
случае процесс начинается после перехода смеси из состояния стекла в переохлажденную жидкость.
В жидкофазных системах в качестве разбавителей ТФЭ испытаны линейные и разветвленный перфторалканы,
перфторароматические соединения, а также (ГФП)2 и тример (ГФП)3, и их смеси.
Показано, что для систем с участием олигомеров ГФП существует порог (по температуре, доле фтора,
и глубине процесса) для относительно мягкого протекания процесса с селективным образованием С2F6.
Выше порога для этих систем и единственно возможным для остальных оказывается жесткий режим, где
наряду с сильными разогревами, большой долей продуктов разрыва С=С связи ТФЭ, и расходом разбавителя
наблюдаются также вспышки и взрывы. Для фторирования ТФЭ в конденсированных средах предложен механизм,
основанный на представлениях об образовании возбужденных продуктов экзотермических элементарных актов,
и их влиянии на кинетику процесса. В частности, сделан вывод о стабилизации комплексов фтора с молекулами
ТФЭ и среды в точках изменения фазового состояния компонентов системы, участвующих с ростом температуры
в зарождении радикалов, в том числе – долгоживущих. Обсуждается их роль в процессах фторирования
перфторолефинов.
КЛЮЧЕВЫЕ СЛОВА: тетрафторэтилен, гексафторпропилен, прямое фторирование, перфторорганические конденсированные среды, механизм реакций, долгоживущие перфторрадикалы
Введение
Фторирование органических соединений молекулярным фтором имеет широкое применение для модификации физических и химических свойств полимеров, а также для получения полимерных и низкомолекулярных соединений [1-13].
Уникальность реакции тетрафторэтилена (ТФЭ) с элементарным фтором связана с высокой активностью обеих молекул в радикальных реакциях, которые определяют перспективность использования этой системы для решения многих практических и научных задач. При взаимодействии фтора и ТФЭ π-связь ТФЭ находится под действием положительного мезомерного эффекта атомов в молекуле фтора, в результате чего эта связь сильно поляризуется, и молекула ТФЭ приобретает свойства, близкие к бирадикалу. Этим объясняется чувствительность активированных фтором молекул ТФЭ к присутствию радикалов вообще [14], и активность в радикально-цепных процессах полимеризации, в частности [15]. В то же время, по практике прямого фторирования органических соединений в различных фазах широко известны непредсказуемые эффекты перегрева реакционной среды. Реакция фтора с большинством органических соединений высоко экзотермична из-за образования С-F связей и может сопровождаться горением и взрывом. Процесс можно обезопасить, проводя его в условиях сильного разбавления инертными разбавителями.
В жидкой фазе процесс фторирования ТФЭ может идти на границе раздела жидкость-газ, или в пузырьках газа, и, не всегда являясь полностью гомогенным, признается опасным. Тем не менее, исследования реакций фторирования в жидкой фазе появились и развивались в русле пионерских работ Merritt'a [16,17], впервые показавших возможность присоединения фтора к олефинам определенного типа. Однако, в противоположность прямым процессам хлорирования и бромирования олефинов, получение 1,2-дифторпроизводных присоединением фтора к двойной связи олефина считалось трудной задачей, и развивались способы обхода путей, использующих фтор [9,18,19]. В [13] было предложено использовать (ГФП)3 в качестве высокоэффективной среды для проведения жидкофазного фторирования непредельных перфторированных соединений элементарным фтором, при этом не было обнаружено следов побочных реакций полимеризации или деструкции (со взрывом) исходного мономера, обычно сопровождающих этот процесс.
Присоединение фтора к неразбавленным перфторолефинам исследовалось в газовой, и в жидкой фазах [12,13,20-23]. Было найдено, что при отсутствии сильных разогревов и взрывов главными продуктами этих реакций являются перфторалканы. Однако, что происходит при разогревах, и каков при этом механизм образования продуктов, установлено не было. Установление механизма является необходимым для более обоснованного выбора способов получения перфторорганических соединений, путем прямого фторирования.
Новый этап в исследовании реакций прямого фторирования перфторолефинов был начат после обнаружения долгоживущих
в жидкости перфторалкильных радикалов (ДР) при фторирования (ГФП)3 [21]. По результатам
ряда работ [20-22, 24-28,] в [24] было установлено их образование в результате присоединения атома
фтора к двойной связи перфторолефинов, в данном случае - олигомеров ГФП. Исследование фторуглеродных
радикалов методом ЭПР-спектроскопии в жидкости, как оказалось [27], имеет некоторые преимущества.
Свободное вращение радикала в жидкости усредняет анизотропию сверхтонкого взаимодействия. Тогда ширина
компонент сверхтонкой структуры спектра ЭПР уменьшается, и можно измерить расстояние между компонентами
и их интенсивность с большей точностью. В жидкости могут быть накоплены большие концентрация ДР для
всестороннего изучения. Соответственно, можно выявить специфические особенности фторирования олигомеров
ГФП, и роль ДР в этом процессе. Факт образования ДР в ходе прямого фторирования олигомеров ГФП свидетельствует
о преобладающем характере цепного механизма среди возможных процессов фторирования ненасыщенной связи
[24].
В настоящей работе подробное исследование особенностей фторирования ТФЭ в жидких смесях
проведено с различными перфторорганическими соединениями, а в стеклообразных – только с олигомерами
ГФП. Основной целью работы является выявление условий реализации безопасного процесса, допускающего
корректную интерпретацию наблюдаемых закономерностей. Это может явиться основой для существенного
развития представлений о детальных механизмах указанных процессов.
Методика эксперимента
Характеристики фторорганических соединений, использованных в работе, приведены в таблице 1. Эти вещества и F2 были производства Пермского филиала ФГУП Российский научный центр "Прикладная химия".
Фторирование жидких смесей ТФЭ с разбавителями и отдельно самих разбавителей проводили в различных реакторах, отличающихся способами подачи в реактор органических компонентов смеси и фтора, а также мерами безопасности.
Фторирование разбавителей типа олигомера (ГФП)3, отличающегося низкой реакционной способностью, проводили в реакторе из нержавеющей стали, снабженном мешалкой и системами подачи фтора и отбора газообразных продуктов. Фтор подавали в реактор с жидким перфторолефином при начальном избытке давления фтора (0.3 - 0.7 МПА), а останавливали реакцию, когда этот избыток уменьшался до 0.01 МПа. Скорость такой подачи фтора в реактор составляла около 25 л/час. Исходную температуру варьировали в диапазоне от 263 К до 343 К.
Для фторирования ТФЭ в смесях с жидкими фторорганическими соединениями использовали реактор (сталь 12Х18Н10Т, объем 5,5л.), оборудованный двумя сифонами для подачи газов (F2 и ТФЭ - барботирование), манометрами для измерения давления каждого из них, термопарой и турбинной мешалкой (n = 300 об/мин). Вариация мольного соотношения газовых реагентов α= F2: ТФЭ (1,05-1,2) достигалась регулированием их скоростей от 25 л/час (1,115 моль/час) до 26,2 л/час (1,17моль/час). Начальная температура в реакторе менялась от 203 до 283 К. Уменьшение риска выхода реакции в свободный объем реактора над жидкостью (иногда со взрывом) достигалось благодаря заполнению этого объема пассивированной медной стружкой.
Фазовые превращения и кинетику фторирования ТФЭ в стеклообразных растворах олигомеров ГФП изучали методом низкотемпературной калориметрии [29] с использованием специальных кварцевых кювет. Определенную навеску перфторолефина помещали в кварцевую кювету и вакуумировали до остаточного давления 0.13 Па при 77 K, затем в подавали в нее определенное количество газообразного фтора. Приготовленную таким образом ампулу помещали в калориметр, где в ходе размораживания такого образца отслеживались все возможные тепловые эффекты.
Динамика образования парамагнитных центров была измерена ЭПР спектрометрией. Образец перфторорганических
соединений или перфторолефина помещали в кварцевую ЭПР ампулу (диаметром 4-5 мм) и дегазировали при
77 К до 10-3 мм, затем газообразный фтор напускали в ампулу. Погрешность при определении
абсолютной концентраций парамагнитных центров составила (20-30)%, относительной - (5-10)%. Нагревание
образцов проводили струей паров азота, выдерживая образцы при каждой температуре эксперимента в течение
8-10 мин. ЭПР измерения проводили при 77 K. Спектры ЭПР регистрировали на малогабаритном радиоспектрометре
PS100.X с автоматической обработкой спектров ЭПР. Автоматическая регистрация осуществлялась с использованием
программы EPRWIN. Теоретические расчеты спектров и их симулирование проводились с использованием
программы EPRTOOLS. Разработчик программ EPRWIN и EPRTOOLS – НПП "Адани", г. Минск.
Хроматографический
анализ газовых и жидких продуктов проводили на приборе ЛХМ. В качестве сорбента использовали смесь
дибутилфталата и вазелинового масла (в соотношении 1:1), нанесенную на Al2O3 (25:100). Длина колонки была 5 м., диаметр колонки - 3 мм. Ток детектора был 130 мА. Скорость газа-носителя
(аргон) при анализе газов была равна 1.0 л./час, а при анализе жидкой фазы - 4.91 л/час.
Результаты и их обсуждения
I. Фторирование ТФЭ в твердофазных перфторорганических (ПФО) средах
Реакционная способность ТФЭ к фтору в твердой фазе рассматривается по результатам последовательно проведенной серии экспериментов в системах фторирования индивидуальных ТФЭ и ГФП и в стеклообразных смесях ТФЭ с димером и тримером ГФП. Для последних также рассмотрены фазовые превращения и реакционная способность к фтору в соответствующих диапазонах температур. Все это может дать дополнительные сведения о механизмах появления и участия долгоживущих радикалов в реакции фторирования ТФЭ.
А). Фторирование ТФЭ и ГФП в кристаллическом состоянии
На рисунках 1,1-1' и 1,2-2' представлены калориметрические кривые размораживания охлажденных до 77К образцов
ТФЭ и ГФП, соответственно, которые позволяют сравнить фазовые состояния и реакционную способность
по отношению к фтору 2-х наиболее простых перфторолефинов при достаточно низких температурах.
На рис. 1,1 приведена калориметрическая кривая размораживания охлажденного до 77 К образца ТФЭ. В
ходе нагревания до комнатной температуры регистрируется эндотермический пик плавления кристаллической
фазы ТФЭ (142К).
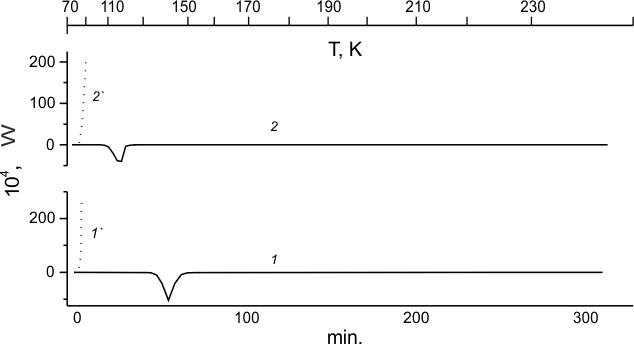
Рис.1. Калориметрические кривые размораживания ТФЭ (1) и ГФП (2), а также смесей ТФЭ:F2=99:1 мол.% (1') и ГФП:F2 = 99:1 мол.% (2').
Из кривой размораживания образца кристаллического ТФЭ с намороженным на его поверхность молекулярным
фтором (см. рис.1,1'), по-видимому следует, что реакция фтора с кристаллическим ТФЭ не происходит
при 77К, но самопроизвольно, и со взрывом, начинается при температуре 85К, являющейся точкой кипения
жидкого фтора.
Полученная таким же образом кривая размораживания кристаллических образцов ГФП
в присутствии фтора (см. рис.1,2') показывает, как и в случае фторирования кристаллического ТФЭ,
что самопроизвольная реакция со взрывом начинается при той же температуре. Заметим, что ГФП имеет
более низкую температуру плавления (115К, рис.4,2), чем ТФЭ, и значительную теплоту плавления ГФП
(-4,9±0,1 кДж/моль). Поэтому для сохранения гомогенности процесса и минимизации энергетических затрат
размораживание этой системы следует останавливать до достижения области плавления ГФП.
Таким образом, получены данные, свидетельствующие об одинаково бурном фторировании кристаллических образцов ТФЭ и ГФП. По-видимому, такое поведение допускает не только радикально-цепной механизм процесса, но и разветвленно-цепной. Как видно, низкотемпературное фторирование перфторолефинов не требует какого-либо инициирования и оно протекает спонтанно. Однако, низкотемпературное хлорирование этих перфторолефинов требует инициирования [30] и оно не происходит спонтанно.
Обнаруженный в настоящих экспериментах бурный режим фторирования кристаллических образцов ТФЭ и ГФП не позволяет установить состав продуктов в случае обоих олефинов. Но тут процесс идет в существенном избытке жидкого фтора над реагирующей частью олефина.
Калориметрические кривые 1' и 2' на рис.1 показывают, что F2 начинает бурно реагировать с кристаллическими ТФЭ и ГФП только при температуре кипения F2. Однако, возможность начала такой реакции нельзя исключить и при температурах ниже 85 К. При используемой низкотемпературной калориметрическая методике F2 намораживается на кристаллический образец при 77К перед началом разогрева образца, Это происходит в блоке калориметра без экспозиции, требуемой для унификации выхода на постоянный режим работы. Поэтому повышение энергии образца за счет реакции может произойти быстрее, чем за счет регистрируемого термопарой программируемого разогрева. Для того, чтобы ответить на вопрос: "Фторируются ли кристаллические ТФЭ или ГФП до температуры кипения фтора?", необходимо провести исследование фторирования ТФЭ и ГФП при температуре жидкого гелия.
Б) Фторирование системы ТФЭ - (ГФП)2 в стеклующейся матрице.
Рассмотрим, как в ходе размораживания образцов, приготовленных при 77К, изменяются фазовые состояния и реакционная способность по отношению к фтору для смеси (ГФП)2 - ТФЭ и каждого из ее компонентов (рис.2).
Фазовое состояние системы (ГФП)2 - ТФЭ
Калориметрическая кривая размораживания чистого (ГФП)2 приведена на рис.2,1. При быстром замораживании до 77К (ГФП)2 полностью переходит в стеклообразное состояние. В ходе размораживания такого образца в калориметре при ~110К наблюдается переход из состояния стекла в переохлажденную жидкость (характерное изменение теплоемкости в виде "ступеньки"). Кристаллизация образца происходит при 124К и плавление при 176К. Фазовое состояние системы (ГФП)2- ТФЭ при 77К показывает его зависимость от концентрации ТФЭ в образце. При концентрации [ТФЭ] >20 моль % эта система представляет собой смесь кристаллического ТФЭ и стеклообразного (ГФП)2. В этом случае при размораживании образца (рис.2,1) регистрируются: эндотермический пик плавления кристаллического ТФЭ, переход (ГФП)2 в переохдажденную жидкость, кристаллизация (ГФП)2, а также плавление (ГФП)2 - при тех же температурах, которые были установлены для индивидуальных фазовых переходов ТФЭ и (ГФП)2. При концентрации [ТФЭ] ≤20 моль% замороженный при 77К образец (ГФП)2-ТФЭ представляет собой прозрачное однородное стекло. Заметим, что для получения полностью стеклообразных растворов (ГФП)2-ТФЭ охлаждение образца следует проводить достаточно медленно (~60 град/мин). При размораживании такого образца в калориметре пик плавления ТФЭ практически не наблюдается (менее 1% кристаллов ТФЭ). Температуры других фазовых превращений такого стеклообразного раствора (ГФП)2-ТФЭ совпадают с таковыми для чистого (ГФП)2 при 77К.
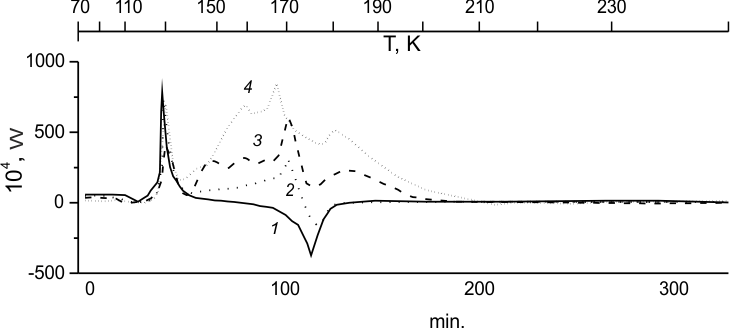
Рис.2. Размораживание (ГФП)2 (1) и смесей (ГФП)2:F2 = 94:6 мол.% (2), (ГФП)2:F2=80:20 мол.% (3) и (ГФП)2: ТФЭ:F2= 53:12:35 мол.% (4).
Фторирование чистого димера (ГФП)2 при низких температурах
Вспомогательная роль (ГФП)2 как более инертного партнера- разбавителя ТФЭ при фторировании, и наличие в нем двойной связи, вызвали необходимость отдельного исследования реакционной способности (ГФП)2 по отношению к фтору.
Рис. 2,2 показывает, что при 77К реакция фтора с (ГФП)2 не происходит. Также никакого тепловыделения не наблюдается при разогреве (ГФП)2 до температур перехода в переохлажденную жидкость (рис. 2,2-3). При дальнейшем повышении температуры появляется область тепловыделения, в целом имеющая характерную колебательную структуру, распространяющуюся от точки кристаллизации (ГФП)2 до температур, достигаемых уже после его плавления. То, что эта область связана именно с фторированием, можно видеть из сравнения калориметрических кривых (рис. 2,2-3), полученных при разных исходных соотношениях α=F2:(ГФП)2. Действительно, локальные изменения тепловыделения на них колеблются вдоль всей области пропорционально α, хотя и в разной степени. Особенно хорошо это видно по максимуму пика, который наиболее близок к точке плавления (ГФП)2. Он наиболее сильно меняется при изменении доли фтора, и менее всего спадает и сдвигается в сторону этой точки с ростом доли фтора в системе.
Полученные данные можно интерпретировать следующим образом. Во-первых, при кристаллизации образца (ГФП)2 может произойти некоторое перемешивание жидкого фтора со стеклообразным димером. Появляются пары фтор - олефин, с ростом температуры образующие молекулярные комплексы с небольшим тепловыделением (первый пик). Его спад определяется поглощением энергии на преодоление барьера взаимодействия фтора с олефином. Весь высокий тепловой эффект этой реакции переходит в колебательную энергию образующегося перфторалкана С6F14, распад которого на радикалы инициирует цепной процесс (второй пик). Спад второго пика связан не только с затратами на плавление образца, но и на преодоление барьера стадии, лимитирующей развитие процесса, идущего уже в жидкой фазе (третий пик). Зависимость тепловыделения за счет реакции (скорость) от недостающего фтора характеризуется порядком, растущим вдоль калориметрической кривой, что говорит об усложнении кинетической системы. Меняется также величина Еэфф.
Низкотемпературное фторирование ТФЭ в стеклующейся матрице димера (ГФП)2.
На рис. 2,4 представлена калориметрическая кривая размораживания (в присутствии фтора) раствора (ГФП)2-ТФЭ, состав которого определяет однородность этого стекла при 77К, содержащего в основном не кристаллический ТФЭ и стеклообразный (ГФП)2, кристаллизующийся при 124К. Наблюдаемое в этом случае тепловыделение, связанное с реакцией, имеет структуру, аналогичную таковой для фторирования стеклообразного чистого (ГФП)2 (рис.2, 2-3), и начинается лишь по достижении зоны кристаллизации (ГФП)2. В то же время нетрудно видеть, что процесс фторирования стеклообразного раствора (ГФП)2-ТФЭ начинается раньше и протекает более интенсивно, чем фторирование чистого (ГФП)2.
Заметим, что относительную интенсификацию реакции в смеси с ТФЭ можно было бы отнести за счет повышения в ней доли фтора в 2 раза. Однако взрыва, характерного для фторирования кристаллического ТФЭ при 85 К (рис. 1,1'), не наблюдается и в этом случае, даже при больших температурах.
Более интенсивный ход фторирования стеклообразного раствор (ГФП)2-ТФЭ, по сравнению с чистым (ГФП)2, можно объяснить, предположив, кроме упомянутой роли комплексов фтора с (ГФП)2, - еще более существенную роль таких же комплексов с ТФЭ. Но поскольку в данном эксперименте (рис. 2,4) для ТФЭ, в отличие от (ГФП)2, кристаллической фазы практически нет, пары ТФЭ – фтор могут появиться во время кристаллизации (ГФП)2. Из-за большей легкости перехода этих пар в комплексы связанный с этой стадией первый пик тепловыделения заметно смещен в сторону меньших температур, начинаясь еще до окончания кристаллизации (ГФП)2. Последующий переход комплексов в сильно колебательно возбужденный продукт присоединения фтора к двойной связи ТФЭ, также имеет меньшую энергию активации. Поэтому связанный с ним спад тоже кончается заметно быстрее, а инициирование цепного процесса при распаде возбужденного перфторалкана (С2F6*) начинается раньше. Также можно констатировать, что в этом случае процесс в наименьшей степени тормозится из-за потерь энергии на плавление (ГФП)2. В то же время, заметное сходство кинетических кривых, соответствующих фторированию чистого (ГФП)2 и его смеси с ТФЭ, связано с тем, что реакция ТФЭ, хотя и начинается раньше, инициируя цепной процесс с участием (ГФП)2, тем не менее, идет параллельно с реакцией (ГФП)2. А этот процесс проходит все описанные выше стадии с участием аналогичных комплексов фтора с (ГФП)2, которых в системе гораздо больше, чем таковых с ТФЭ.
Полученные данные позволяют объяснить, почему кривая размораживания для смеси (ГФП)2-ТФЭ (рис.2,4) не фиксирует сколько-нибудь заметного тепловыделения, связанного с фторированием ТФЭ в области 85К, где для кристаллического образца ТФЭ наблюдался взрыв (Рис.1,1'). А также - почему в системе со смесью (ГФП)2- ТФЭ не происходит взрыв при больших температурах и концентрациях фтора. Из этих данных можно предположить, что причиной взрыва при низкотемпературном фторировании ТФЭ явилось сочетание высокой упорядоченности кристалла с повышением подвижности фтора в точке кипения, так как именно это могло способствовать как образованию комплексов фтора с ТФЭ, так и всей связанной с ними эволюции процесса, описанной выше. Такой путь не может реализоваться при фторировании ТФЭ в смеси с димером ГФП, так как, прежде всего, в этой смеси практически отсутствует кристаллическая фаза ТФЭ. Поэтому реализуется путь, связанный с кристаллической фазой (ГФП)2 , на которой и формируются упорядоченные пары не только фтор-(ГФП)2, но и фтор - ТФЭ. Что касается отсутствия взрыва при фторировании смеси, то, не считая эффекта сильного разбавления ТФЭ в димере, следует еще учитывать рассмотренный уже для аналогичных реакций в жидкой фазе эффект ингибирования процесса фторирования ТФЭ разбавителем, который связан с поглощением ведущих цепь атомов фтора как самим димером, так и получающимися из него долгоживущими радикалами ДР(1) и ДР(2) (см.ниже).
В) Низкотемпературное фторирование ТФЭ в стеклующейся матрице (ГФП)3
Фазовое состояние низкотемпературной системы ТФЭ - (ГФП)3
Чистый (ГФП)3 при быстром замораживании до 77 К полностью переходит в стеклообразное состояние. Переход из этого состояния в переохлажденную жидкость наблюдается при Тс~150К ("ступенька"), после чего переохлажденная жидкость переходит в термодинамически стабильную - без кристаллизации, и, соответственно, без плавления (рис.3, 1).
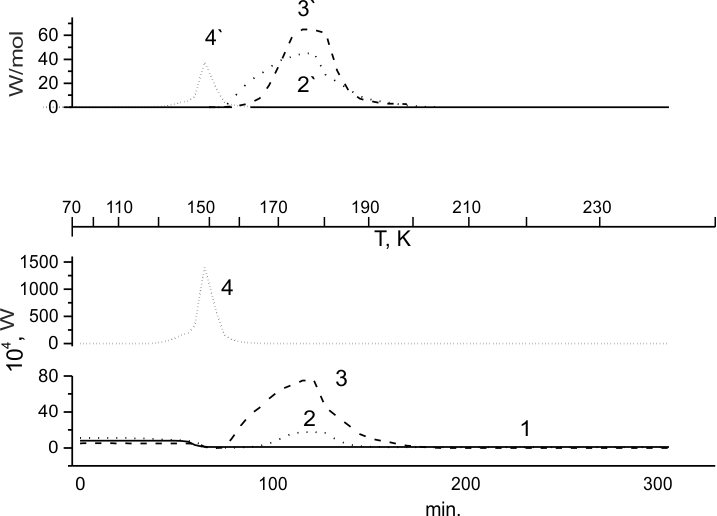
Рис.3. Размораживание (ГФП)3 (1), и смесей (ГФП)3:F2= 99:1 (2), (ГФП)3: ТФЭ: F2 = 85.5:10:4.5 мол.% (3) и (ГФП)3: ТФЭ = 48:52 мол.% (4). Кривая "4" получена для образца "4", предварительно облученного дозой 140 кГр гамма – лучами 60Со при 77 К. Интегральные тепловыделения 2', 3' и 4' регистрируемые на калориметрических кривых размораживания образцов 2, 3 и 4, соответственно.
Исследование фазового состояния смесей (ГФП)3-ТФЭ при 77К, как и в случае с (ГФП)2, показало его зависимость от концентрации ТФЭ в образце. При [ТФЭ]>60 моль % эта система представляет собой смесь кристаллического ТФЭ и стеклообразного (ГФП)3, тогда как при [ТФЭ]<60 моль % она полностью стеклообразна. Поэтому калориметрическая кривая размораживания смеси (ГФП)3- ТФЭ, содержащей 52 мол. % ТФЭ (рис.3,4), аналогична таковой для чистого образца (ГФП)3 (рис. 3, 1), и отвечающая ему "ступенька" перехода в переохлажденную жидкость также фиксируется при температуре ~150К.
Кинетика низкотемпературного фторирования (ГФП)3
Ранее была показана заметная активность двойной связи (ГФП)3 в низкотемпературной реакции присоединения фтора [24,29,31]. На рис. 3, 2 представлена калориметрическая кривая размораживания (ГФП)3 в присутствии 1% фтора. Из нее видно, что при 77 К реакция фтора со стеклообразным (ГФП)3 не происходит. Заметное тепловыделение также не наблюдается при разогреве образца (ГФП)3 в присутствии F2 в области "ступеньки" - при переходе системы в переохлажденную жидкость. Область тепловыделения, связанная с реакцией, становится отличной от теплового фона только по достижении температуры ~160 К. Однако по удельному тепловыделению в этой области можно заметить, что хотя оно и мало, и заканчивается полностью уже при температуре ~190 К, но его начало приходится как раз на период, начинающийся сразу после перехода типа "ступенька".
Низкотемпературное фторирование стеклообразной смеси ТФЭ - (ГФП)3.
Из калориметрических кривых на рисунках 3,2 и 3,3 можно видеть, что добавление ТФЭ в стеклообразную матрицу
(ГФП)3 увеличивает скорость (тепловыделение) процесса фторирования, как это наблюдалось
и для (ГФП)2
В то же время, сравнивая калориметрические кривые на рис. 2,4 и 3,3,
демонстрирующие ход фторирования для стеклообразных смесей ТФЭ с (ГФП)2 и (ГФП)3,
соответственно, можно видеть, что в последнем случае скорость фторирования ниже, чем в первом, более
чем на порядок.
Из сказанного выше о фторировании чистого (кристаллического) ТФЭ, и его стеклообразных смесей с перфторолигомерами ГФП, можно сделать предварительные выводы.
По-видимому, механизм фторирования ТФЭ вообще, и стадия образования комплексов [F2...F2C=CF2], в частности, играют важную роль в процессе фторирования не только самого ТФЭ, но и перфторолефинов, содержащихся в системе. Наличие комплекса облегчает ассоциацию молекул фтора с ТФЭ, приводящую к образованию высоко возбужденных молекул перфторэтана и их распаду на первичные радикалы. Но в случае с ТФЭ такая ассоциация протекает с минимальной для данного ряда олефинов энергией активации, что, соответственно, сдвигает начало процесса в сторону относительно более низких температур. Между тем, образование и стабилизация такого комплекса зависит от фазового состояния и природы матрицы, используемой при низкотемпературном фторировании ТФЭ. Как было рассмотрено выше, такой комплекс может стабилизироваться в ходе кристаллизации (ГФП)2 из переохлажденной жидкости, а выделяющееся при этом тепло может эффективно активировать переход стабильного комплекса в пару реагирующих молекул. Поэтому фторирование ТФЭ в матрице (ГФП)2 протекает относительно эффективно.
В отличие от стеклообразного (ГФП)2, матрица (ГФП)3 не кристаллизуется. Тем не менее, фторирование (ГФП)3 в присутствии ТФЭ протекает быстрее, и начинается раньше, а именно - сразу после фазового перехода 2-го рода (скачкообразное уменьшение теплоемкости при переходе стекла в переохлажденную жидкость). Возможно, что и в этих условиях нельзя исключать стабилизацию комплекса [F2...F2C=CF2], и последующее зарождение радикалов через него, так как в этом случае система оказывается в область достаточно высоких температур.
В таблице 2 приведены данные по теплоте реакции фторирования перфторолефинов. Анализ полученных данных свидетельствуют что присоединения фтора по карбенному механизму может происходит если ТФЭ образует свою собственную кристаллическую фазу (взрыв!). Теплота реакции фторирования стеклообразных растворов ТФЭ-(ГФП)2или ТФЭ-(ГФП)3 составляет 440 – 528 кДж/моль. Поскольку, реакция фторирования молекул растворителей (ГФП)2 и (ГФП)3 накладывается на реакцию фторирования молекул ТФЭ, оценить величину удельной теплоты фторирования и длину кинетической цепи фторирования ТФЭ не удается.
Таблица 2. Теплоты реакций фторирования перфторолефинов
| Состав реагентов, (мол%) | *Теплота реакции, кДж/моль | ||
| Перфторолефин |
F2 |
ТФЭ |
|
|
ТФЭ (99.2) |
0.8 |
Взрыв |
|
|
ГФП (99) |
1 |
Взрыв |
|
|
(ГФП)2 (80) |
20 |
528 |
|
|
(ГФП)2 (94) |
6 |
450 |
|
|
(ГФП)2 (53) |
35 |
12 |
507 |
|
(ГФП)3 (99) |
1 |
470 |
|
|
(ГФП)3 (85,5) |
4.5 |
10 |
518 |
|
(ГФП)3 (48) |
52 |
169** |
|
|
CF3-C6F4-CF3 (71) |
29 |
440 |
|
|
CF3-C6F4-CF3 (54) |
46 |
500 |
*-Теплота реакции была определена относительно количества израсходованного фтора
(ГФП)3 после их гамма – облучения при 77 К дозой 140 кГр
**- Теплота полимеризации (кДж на моль израсходованного
ТФЭ) была определена из калориметрических кривых размораживания стеклообразного раствора ТФЭ в (ГФП)3
после их гамма – облучения при 77 К дозой 140 кГр
II. Фторирование ТФЭ в среде жидких перфторорганических соединений.
А) Состав продуктов.
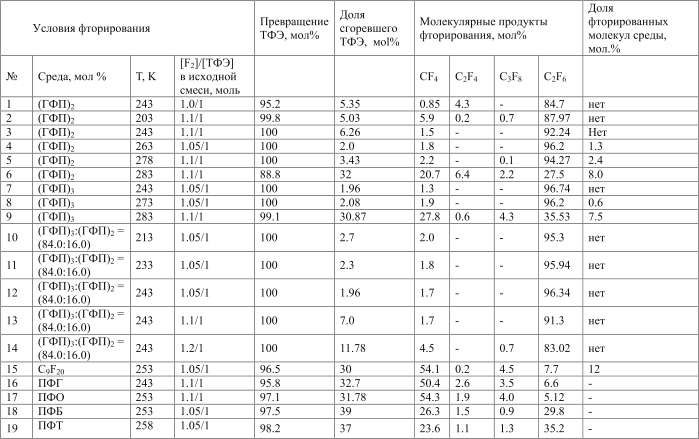
Состав продуктов фторирования ТФЭ в различных жидких ПФО средах по данным хроматографического анализа для различных условий представлен в таблице 3.
Из таблицы видно, что при относительно низких температурах и соотношениях F2 : ТФЭ основным продуктом прямого фторирования ТФЭ – как в среде олигомеров ГФП, так и в их смесях, является C2F6. На основании этих данных можно предположить, что механизм мягкого фторирования ТФЭ, в основном, приводит к насыщению его двойной связи, обеспечивая высокую селективность! процесса по C2F6. Можно также полагать, что выход заметных количеств других молекулярных продуктов (CF4 , C3F8 ... ) в этих же средах может быть связан с деструкцией ПФО среды.
Данные табл. 3 по поведению систем фторирования ТФЭ в средах линейных (ПФГ, ПФОК) и разветвленного (C9F20) перфторалкана, а также перфторароматических (ПФБ, ПФТ) соединений в целом показывают далеко не селективный выход C2F6 при заметно меньших температурах, чем в опытах 6 и 9 в среде (ГФП)3. Так, в среде перфторароматических соединений, несмотря на то, что C2F6 составляет около половины всех молекулярных продуктов фторирования ТФЭ, с большим выходом (38.5% - 45%) образуется также CF4. Все это говорит о более жестком режиме протекания процесса. Кроме того, в этих случаях, за исключением системы с перфторалканом C9F20, вообще нет признаков заметной границы мягкого и жесткого режимов (по температуре и соотношению F2 : ТФЭ). Но и здесь, где состав продуктов не меняется в интервале температур ~210-250К, имеется значительная доля продукта CF4, считающегося признаком деструкции ПФО среды. На температурном пределе мягкого фторирования в системе с C9F20 (253К) перфторметан является основным продуктом, а C2F6 составляет всего 12.5 мол.%. Но если у олигомера имеется двойная связь, то полностью насыщенная молекула C9F20 (идентичная продукту насыщения фтором двойной связи (ГФП)3) не располагает никакой активностью, возможно, кроме моментов вращения в местах связи третичного углерода с СF3 группами. Заметим, что это происходит на фоне эффективного расходования молекул ТФЭ.
Б) Горение, наблюдаемое при прямом фторировании ТФЭ в среде жидких перфторорганических соединений.
Из наших экспериментов по фторированию в среде перфторированных жидкостей можно сделать заключение о возможности реакции части молекул TФЭ над жидкостью в газовой фазе, что часто демонстрируется возникновением вспышек, или даже взрывов. В результате в реакторе появляются следы «горения» (сажа, смолы, и другие неполезные продукты), выход которых не поддается точным измерениям. Тем не менее, и без протекания газофазной реакции критерием эффективности процесса фторирования можно считать минимальный выход таких продуктов. Одной из гарантий правильного подбора эффективной среды фторирования ТФЭ является контролируемый расход этого реагента с учетом образования молекулярных продуктов. Долю прореагировавших молекул ТФЭ, не найденную среди хроматографически идентифицированных газообразных продуктов, и суммированную с продуктами, для получения которых предположительно требуется деструкция С=С связи ТФЭ, мы условно относим к продуктам "горения". Для этой доли на рис. 4 и 5 приведены зависимости от температуры и соотношения концентраций F2: ТФЭ, соответственно для всех исследованных сред, и для наиболее оптимальной смеси [(ГФП)2] : [(ГФП)3] =16 : 84 ( mol.%) при комнатной температуре. При всей условности такого деления рис.4 и 5 показывают качественное разделение типов поведения исследуемых систем при достаточно широкой вариации параметров. Рис.6 демонстрирует возможность "горения" при фторировании одного из наиболее инертных разбавителей - (ГФП)3.
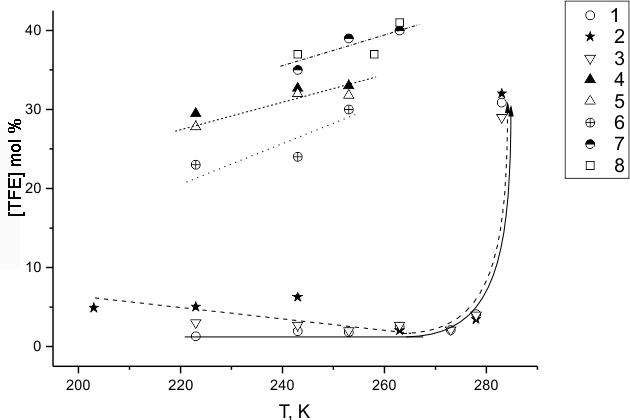
Рис.4. Зависимость доли молекул ТФЭ, участвующих в «горении», от температуры фторирования ТФЭ в среде (ГФП)3 (1), (ГФП)2 (2), смеси [(ГФП)2]:[(ГФП)3]=16:84 (3), н-C6F14 (4), н-C8F18 (5), C9F20 (6), ПФБ (7) и ПФТ (8).
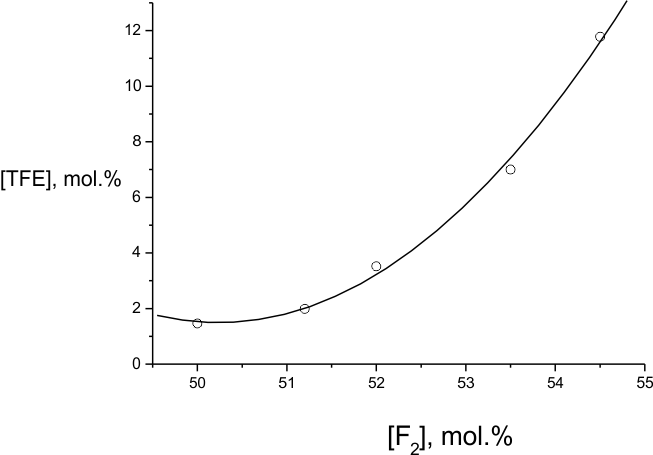
Рис.5. Зависимость доли молекул ТФЭ, участвующих в «горении», от исходного соотношения F2:ТФЭ при фторировании в смеси [(ГФП)2]:[(ГФП)3]=16:84 мол.%. Температура фторирования 303 К.
Из рис.4 видно, что примерно одинаково спокойное поведение систем фторирования ТФЭ в среде олигомеров ГФП и их смесей, и отчасти - перфторалкана C9F20, наблюдается на более или менее протяженном интервале относительно низких температур. Гораздо более бурное поведение эти же системы проявляют при температурах выше некоторой пороговой, характерной для каждой из них. Рис.5 для системы со смесью [(ГФП)2]:[(ГФП)3]=16:84 (mol. %) также показывает пороговый характер влияния концентрации фтора на режим процесса. Можно констатировать, что увеличение начальной температуры действует так же, как повышение скорости процесса при повышении исходной концентрации фтора. Поскольку концентрация ТФЭ в этих экспериментах постоянна, можно оценить, что порядок скорости реакции по фтору меняется примерно от 1 до 2 с ростом доли фтора. Это может означать, что процесс при меньших долях фтора контролируется зарождением по молекулярному взаимодействию фтора с ТФЭ, а с ростом доли фтора – еще и взаимодействием фтора с одним из радикалов. Эта реакция, равно чувствительная к повышению температуры и доли фтора, по-видимому, лимитирует разветвление цепей, и мы полагаем, что речь может идти о реакции F 2 с дифторкарбеном :CF2.
Положение системы с разветвленным перфторалканом C9F20 на рис.5 является промежуточным между теми, которые имеют порог мягкого режима, и которые при минимальных температурах ведут себя бурно. Среди последних, наряду с активно фторирующимися перфторароматиками, находятся также линейные перфторалканы (н-C6F14 и н-C8F18). Это весьма прочные молекулы, прямое фторирование которых исключено. Поэтому их влияние на начальный процесс должно быть так же мало, как и в случае разветвленного перфторалкана C9F20, и во всех трех случаях – меньше, чем влияние (ГФП)3, которое может быть даже ингибирующим. Это может быть причиной изначально высокой скорости фторирования ТФЭ в среде перфторалканов.
Из данных табл. 3 для всех разбавителей, слабо влияющих на начальный процесс фторирования ТФЭ, видно, что они разлагаются при глубокой степени превращении последнего [в случае линейных перфторалканов проверка на их разложение в результате реакции TFE не проводилась из-за высокой начальной скорости, близкой к взрыву]. Разбавитель (ГФП)3, кроме того, расходуется с ростом температуры в системе собственного фторирования рис.6. Однако это происходит при более высоких температурах, чем это имеет место в присутствии ТФЭ, что говорит о роли реакции самого ТФЭ, как первичного процесса, приводящего к деструкции более инертного разбавителя. Возможно, что более глубокая стадия этого процесса сопровождается столь большими тепловыми эффектами, что прочные молекулы указанных разбавителей способны деструктировать благодаря эффективному V-V обмену энергии с колебательно возбужденными продуктами горения ТФЭ. Судя по составу продуктов реакции в соответствующих случаях (см. табл. 3 N 6,9,15-17), где большая часть продуктов может получиться из радикалов ·CF3, речь может идти также о разрыве С-С связи молекул алканов. Заметим, что продуктами насыщения С=С связи обоих олигомеров ГФП являются перфторалканы.
Если деструкция перфторалканов в самом деле приводит к образованию радикалов ·СF3, то их фторирование дает дополнительные количества свободных атомов фтора, удлиняя цепь фторирования основного реагента ТФЭ, и внося свой вклад в накопление наблюдаемых продуктов, наряду с процессами насыщения или расщепления двойной связи молекул ТФЭ.
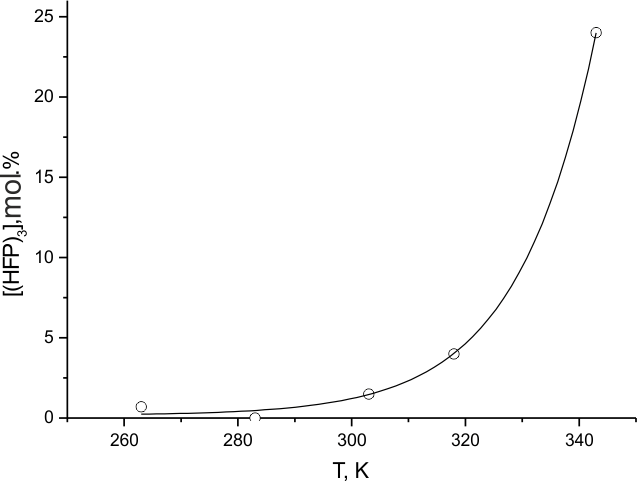
Рис.6. Зависимость доли деструктирующих молекул (ГФП)3 от температуры собственного фторирования.
В) Исследование фторирования тримера ГФП в жидкой фазе..
Показанные выше преимущества фторирования ТФЭ в среде жидкого тримера (ГФП)3 заставили более
подробно исследовать фторирование чистой жидкости (ГФП)3, изучавшееся ранее в [20-22,
32,] методами ЭПР - спектроскопии.
Все эксперименты при температуре не выше комнатной показали,
что более 96% молекул (ГФП)3 превращается в разветвленный перфторалкан C9F20.
На рис. 7. и 8. представлены зависимости скорости фторирования (ГФП)3 от температуры и
давления фтора. Как и следовало ожидать, рост обоих параметров приводит к росту скорости. Однако,
заметная на рис.7 крутизна кривой 2 по сравнению с кривой 1 может означать, что рост давления фтора
и температуры благоприятствуют добавлению в механизм процесса стадии, лимитирующей развитие цепей,
и эта стадия имеет большее значение энергии активации, чем таковая для захвата атома фтора С=С связью
(ГФП)3 .
Это согласуется с ходом зависимости скорости реакции от давления фтора (рис. 8) при Тком. Также, сравнение данных рис. 7, 8 и таковых для температурной зависимости доли молекул (ГФП)3, подвергшихся деструкции при фторировании этого вещества (рис.6), подтверждает сделанный раньше вывод о том, что преимущественное превращение молекул в насыщенный продукт C9F20 возможно только в ограниченной области низких температур и концентраций фтора. За пределами этой области реализуется другой механизм реакции, где активное участие могут принимать частицы, образовавшиеся при деструкции С=С связи.
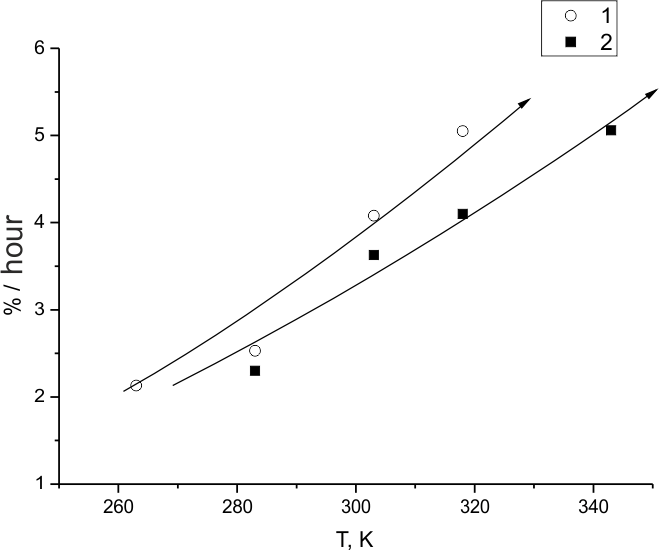
Рис.7. Зависимость скорости фторирования (ГФП)3 от температуры при 0.5 МПа (1), и 0.3 МПа (2)
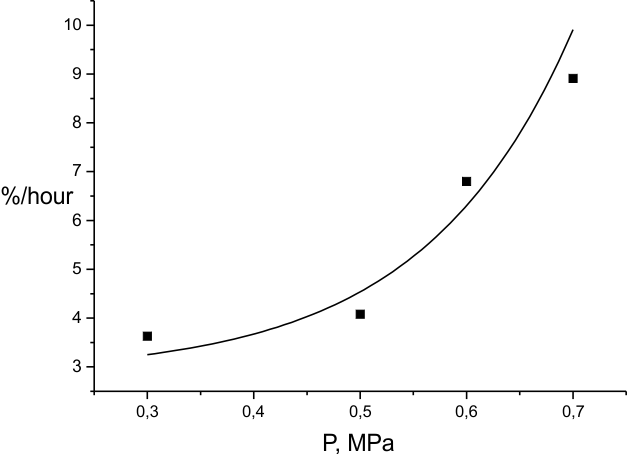
Рис.8. Зависимость скорости фторирования (ГФП)3 от давления фтора при 303К.
В работе [12] было показано, что среди продуктов деструктивного фторирования (ГФП)3 наблюдаются такие, как CF4 и C3F8, что может объясняться взаимодействием соответствующих радикалов (·CF3 и ·C3F7) с молекулярным фтором. Указанные продукты образуются с большим запасом внутренней энергии, которая, однако, может только передаваться среде с повышением ее температуры. Это ситуация, типичная для реализации цепно-теплового взрыва. И действительно, при начальных температурах выше 343К фторирование (ГФП)3 сопровождается постоянными вспышками или горением.
Полученные результаты свидетельствуют, что отсутствие взрывов при фторировании ТФЭ в среде (ГФП)2, (ГФП)3 и их смесях (в определенной области относительно низких температур), очевидно, не является результатом только разветвленной структуры этих веществ и связано с наличием в них двойной связи, поглощающей свободные атомы фтора, ведущих цепь фторирования ТФЭ. Замедленный таким образом, этот процесс за те же времена не получает развития, достаточного для осуществления горения. То есть, олигомеры ГФП можно считать наиболее инертными разбавителями фторирования ТФЭ в обозначенном диапазоне температур.
III. Образование и поведение ДР в жидкой и твердой фазах при фторировании чистых олигомеров (ГФП)2 и (ГФП)3, и в присутствии ТФЭ
А) Кинетика накопления ДР в ходе фторирования стеклообразных (ГФП)2 и (ГФП)3
Полученные данные по регистрации тепловыделения в ходе размораживания фторирующихся образцов (ГФП)2 (рис.2, 2,3) и (ГФП)3 (рис.3, 2), а также каждого из этих образцов с добавлением ТФЭ (рис.2,4 и рис.3,3, соответственно) интересно сравнить с ЭПР исследованиями ДР, образование которых происходит примерно в таких же условиях [24, 31].
В системе фторирования чистых образцов (ГФП)2 зарегистрированы два дублета с расщеплением 18,1 и 5,28 мТл, приписанные радикалам типов ДР(1) и ДР(2), соответственно. В отношении ДР(1) было предположено, что эти радикалы образуются в результате присоединения атома фтора к двойной связи (ГФП)2. Происхождение ДР(2) не обсуждалось. Зависимости концентраций радикалов ДР(1) и ДР(2) от температуры показаны на рис 9,а, 1,2. Видно, что радикалы обоего типа образуются сразу после кристаллизации (ГФП)2 (125К), и хотя их максимальные концентрации заметно отличаются, ход их роста с температурой, по крайней мере до середины максимума (~170К), можно считать симбатным. Эта часть кривых накопления ДР-1 и ДР-2 соответствует той части калориметрических кривых (рис.2, 2-4), которая предшествует зоне плавления и где тепловыделение сильнее всего растет в максимуме с повышением доли фтора. Дальнейший рост ДР-1 происходит как в области плавления образца (ГФП)2, так и в жидкой фазе, а спадает после максимума (~215К) в условиях, когда тепловыделение вообще не фиксируется. Это позволяет однозначно связать природу накопления и гибели радикалов ДР(1) с началом и окончанием фторирования (ГФП)2, тогда как накопление ДР-2 - лишь с началом этого процесса.
Касаясь природы возникновения радикалов ДР(2) в системе фторирования (ГФП)2, необходимо обратить внимание на то, что образующиеся в ней радикалы ДР(2), и ДР(3), наблюдаемые при фторировании (ГФП)3 (рис. 9,б,3), близки как по параметрам спектров ЭПР [24, 31], так и по времени жизни, что может быть указанием на их аналогичную структуру. В соответствии с начальной кинетикой накопления радикалов ДР(1) и ДР(2), можно полагать, что они могут образоваться одновременно - при зарождении. То есть, распад перфторалкана, химически активированного в реакции экзотермического присоединения молекулярного фтора к (ГФП)2, именно в твердой фазе из-за большого времени его жизни может произойти по двум каналам - с образованием 2-х пар радикалов, в одной из которых атом фтора и радикал-·C6F13, (ДР-1), а в другой -·CF3 и радикал ·C5F11. Радикал ·CF3 фторируется молекулярным фтором, повышая концентрацию свободных атомов фтора, присоединяющихся к С=С связи олефина. Поэтому радикалы ДР-1 образуются не только при зарождении, но и в цепном процессе. До конца относительно низкая, но практически постоянная концентрация ДР-2, может являться свидетельством того, что эти радикалы не являются конкурентными компонентами в системе фторирования.
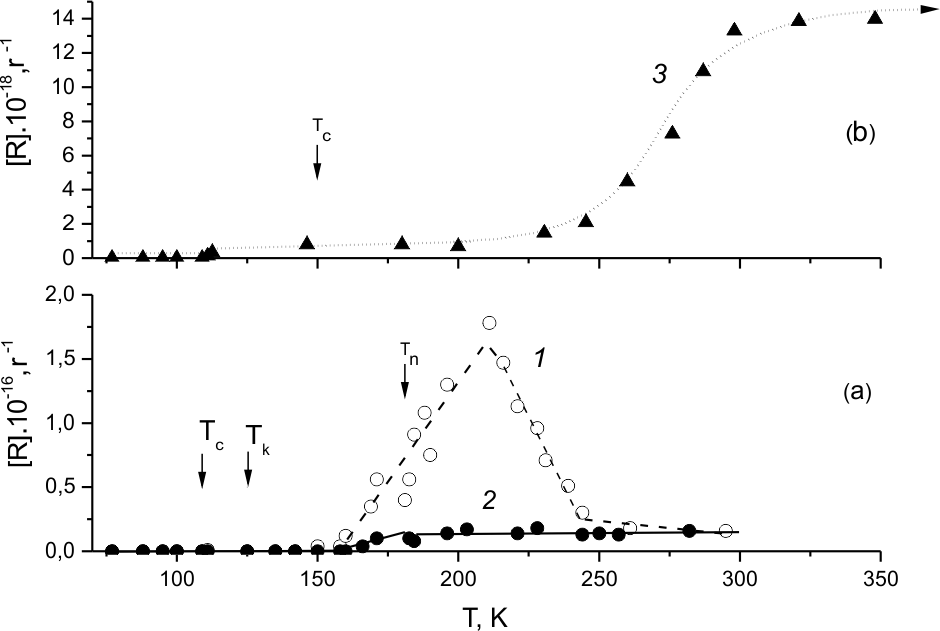
Рис.9.а Концентрации ДР(1)-1 и ДР(2)-2 в ходе разогрева образца (ГФП)2, при 4.5 мол % фтора.
Рис.9.б Концентрации ДР(3)-3 в ходе разогрева образца (ГФП)3, при 3 мол % фтора.
В спектрах ЭПР фторированного (ГФП)3 [20-22,24,31,32] был обнаружен (и приписан радикалам ДР(3)) долгоживущий дублет с плохо разрешенным расщеплением 4,4 мТл, зафиксированным при концентрациях этих частиц больше 1018г-1. В этом радикале, как и в ДР(2), сверхтонкое взаимодействие (СТВ) неспаренного электрона с четырьмя β-атомами фтора не эквивалентно из-за заторможенного вращения - в данном случае перфторэтильной и перфторизопропильной групп. Поэтому в спектре ЭПР ДР(3) при 77К регистрируется не квинтет, а дублет от СТВ неспаренного электрона с одним β-атомом фтора [24,31]. На полувысоте этот плохо разрешенный дублет имеет ширину ~8.0 мТл.
Зависимость выхода от температуры на рис.9, б, 3 получена при проведении фторирования (ГФП)3специально для анализа спектров ЭПР. Сравнивая это с термограммой (рис.3,2), можно видеть, что образование радикалов ДР(3) начинается еще при ~115 К, где нет заметного тепловыделения за счет реакции. Наблюдаемая в этих условиях концентрация ДР(3), равная 1*1018г-1, почти не меняется до более чем 200К. Однако при дальнейшем росте температуры наблюдается близкая к экспоненте резкая зависимость концентрации ДР (3) от температуры, и начиная с Тком их количество~1,4*1019г-1 постоянно.
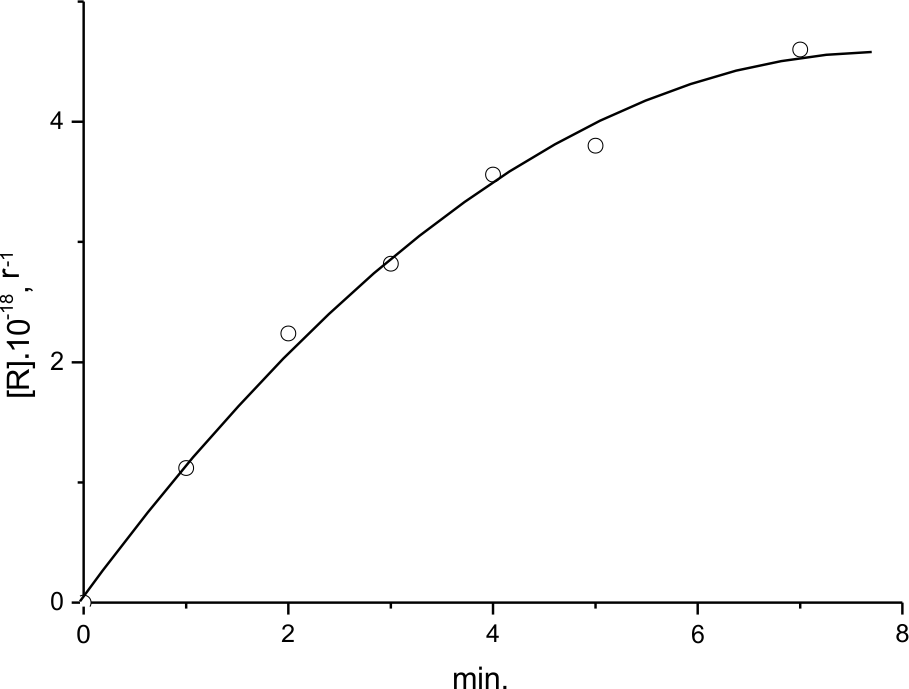
Рис.10. Кинетика накопления ДР(3) в ходе фторирования жидкого (ГФП)3 при 300К.
Из представленных данных можно сделать вывод, что хотя в системе (ГФП)3-F2 цепное присоединение фтора, по-видимому, сильно вырождено, но оно всё же идет. В самом деле, как на стадии зарождения, так и в первом же акте присоединения атома фтора к олефину образуются не реакционноспособные радикалы ДР(3). В то же время, из литературы известно, и в пункте Б) раздела 2 рассматривается возможность образования и участия в процессе фторирования радикалов ·СF3, образующихся при термическом распаде ДР(3). Для самого распада ДР(3) требуется, однако, более значительная активация, чем та, которая достигается в области температур, где наблюдается тепловыделение за счет реакции (160-190К). Возможно, что недостающую для распада энергию (колебательную) эти радикалы могут получить, когда они образуются в результате присоединения атомов фтора к двойной связи тримера. В отличие от тех ДР(3), которые образуются в молекулярной реакции, обозначим их ДР(3)'. Тогда весьма вероятно, что скорость процесса в целом и связанные с ним тепловыделение и концентрации атомов фтора будут малы при низких температурах, но будут расти с ростом температуры по экспоненте, соответствующей энергии активации реакции фторирования радикалов ·СF3, образующихся при распаде ДР(3)' , и дающие развитие цепному процессу через рост концентрации атомов фтора:
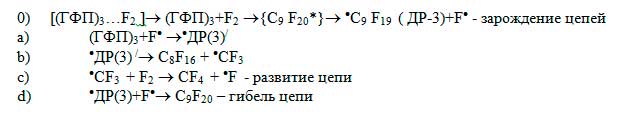
Дополнив эту схему реакцией типа а), приводящей к образованию радикалов ДР(3)', не способных распадаться, мы получим 2 источника радикалов ДР(3), неотличимых от первичных, образующихся при зарождении. Добавим, что доля продуктов, образование которых связано с выделением тепла, невелика, как невелика и концентрация его носителей - атомов фтора.
Б) Кинетика накопления ДР в ходе фторирования чистого (ГФП)3 и в присутствии TФЭ при комнатной температуре.
Образование ДР в результате фторирования олигомеров ГФП, зафиксированное в работах [20-22,24,31,32], без сомнения связано с раскрытием двойной связи в этих молекулах. Можно было ожидать, что заместители при их двойной связи будут оказывать влияние на скорость фторирования и эффективность образования ДР в этом процессе. Для проверки смесь трех изомеров (ГФП)3 общей формулы: (CF3)2CFCR1=CR2CF3, где
R1= C2F5, R2 = CF3 (A)
R1= (CF3)2CF,
R2 = F (Б)
R1=F, R2 = CF2CF2CF3 (В)
фторировалась при комнатной температуре. Картина этого процесса не совсем ясна. Так, концентрации
изомеров Б и В убывают в результате фторирования, а концентрация А не убывает, и даже немного возрастает.
Это значит, что изомеры Б и В более эффективно расходуются при фторировании. Между тем, долгоживущие
в жидкости радикалы одинакового строения [(CF3)3CF]2C*C2F5,
по данным [21], могут образоваться только при фторировании изомеров А и Б. Их ЭПР спектр представляет
собой дублет с расщеплением 4.6 мТл [20], идентичный с таковым для радикалов ДР(3), обнаруженных
в низкотемпературной системе фторирования изначально твердых образцов (ГФП)3. Возникновение
и гибель этих ДР при фторировании (ГФП)3 в жидкой фазе можно представить аналогично схеме
0)-d) для твердо-фазного механизма (см. А, раздел III), как насыщение его С=С связи путем присоединения
к ней молекулярного или атомарного фтора:
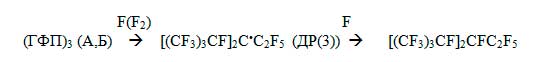
Кинетика накопления ДР(3) в ходе фторирования (ГФП)3 при комнатной температуре представлена на рис. 10. Этот эксперимент был проведен так же, как при получении данных рис.9б,3. Однако в этом случае запись спектров проходила не после размораживания фторирующегося образца до определенной температуры, в том числе, до комнатной, а путем прямой записи спектров ЭПР при Т=300К в течение определенного времени - фактически до тех пор, пока концентрация радикалов перестанет изменяться. По данным В) раздела I для наблюдения кинетики по радикалам выбраны условия, когда деструктивный путь превращения олигомера приторможен. Сопоставляя скорость расхода фтора с кинетикой накопления ДР, тем не менее, можно сделать вывод, что всего фтора расходуется ~ в 5 раз больше, чем идет на образование ДР (около 15% исходной концентрации фтора). Также можно оценить, что в данном эксперименте ~ 30% ДР сохраняются после реакции.
Сделанные оценки исходят из следующих предположений о механизме появления и участия ДР при фторировании (ГФП)3 в жидкой фазе, во многом вобравшим в себя черты механизма в твердой фазе: 1) ДР появляются как в молекулярном взаимодействии, так и в результате присоединения атомов фтора к С=С связи олефина; 2) взаимная рекомбинации промежуточных ДР отсутствует; 3) реакционная способность ДР по отношению к молекулярному фтору маловероятна; 4) не исключено, что единственно возможной реакцией фторирования ДР является рекомбинация с атомом фтора. Одним из доказательств этого является то, что в присутствии (ГФП)3 скорость цепного фторирования молекул TФЭ и вероятность их "горения" снижается, особенно в области температур меньше Тком , о чем уже шла речь выше.
Возможность образования ДР в ходе фторирования TФЭ в среде (ГФП)3 также была исследована. Если газообразный фтор подается при 300 К прямо в ЭПР–ную ампулу, содержащую жидкий (ГФП)3 тогда происходит взрыв. Поэтому, фторирование TФЭ в среде жидкого (ГФП)3 проводилось путем барботирования потоков газообразных TФЭ и F2 сквозь жидкий (ГФП)3.
В условиях такого эксперимента, молекулы как TФЭ, так и (ГФП)3, могут реагировать с молекулами фтора. После реакции жидкий продукт фторирования переносился в ЭПР ампулу, и ДР, образовавшиеся при фторировании, исследовались методом ЭПР. В спектрах ЭПР жидкого продукта такой реакции регистрируется небольшая концентрация ДР. Так, концентрация ДР, образующихся при барботировании ТФЭ (50 мол.%) и F2 (50 мол.%) в среде жидкого (ГФП)3, более чем на четыре порядка меньше, чем таковая в отсутствии ТФЭ. В этом случае очевидно, что фторируется в основном ТФЭ, а (ГФП)3 и ДР поглощают атомы фтора.
Долгоживущие радикалы ДР(3) являются химически инертными при комнатной температуре и не реагируют с молекулярным кислородом или с молекулами TFE. Однако, при фотолизе (λ < 300 нм [33,34]) или нагреве (T>377 К [20]), C-C связи ДР(3) разрываются и образуются активные CF3* радикалы:
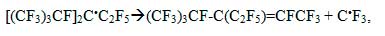
которые могут инициировать полимеризацию ТФЭ или других мономеров [29] :
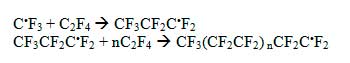
Поэтому, эффективная полимеризация ТФЭ в растворе (HFP)3 в присутствии ДР(1) протекает только в температурной области распада ДР(1) (кривые 1 и 2 на рис. 11). Следовательно, предложенный в [13] механизм передачи атома фтора от долгоживущего радикала C9F19* к радикалам к радикалам CF3CF2* является мало вероятным в ходе фторирования ТФЭ в среде (HFP)3 при комнатной температуре.
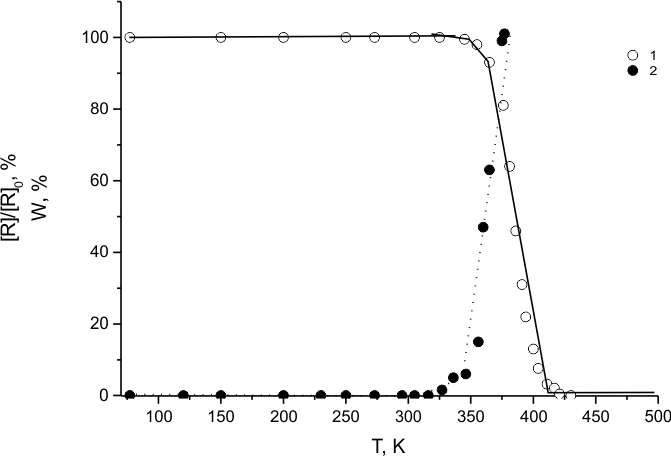
Рис.11. Зависимости концентрации ДР(3) и выхода полимера при полимеризации ТФЭ от температуры в ходе нагрева ТФЭ в среде (ГФП)3 содержащей [ДР(3)] = 2*1018 в 1 грамме (ГФП)3.
Таблица 1. Фторорганические соединения, использованные в работе (нажмите на изображение для загрузки в pdf)
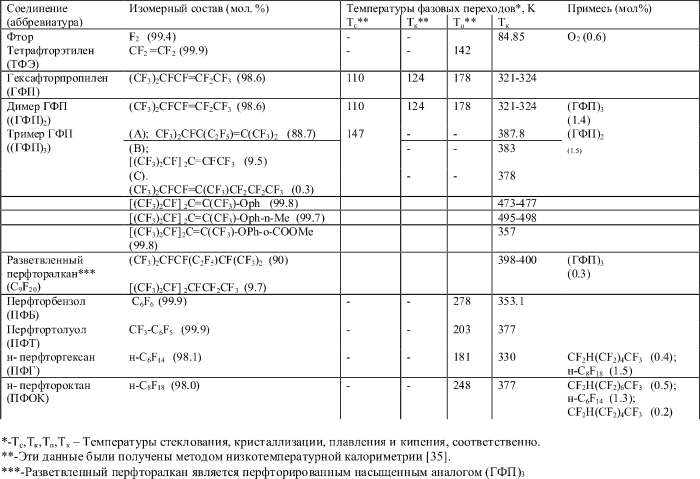
Таблица 3. Молекулярные продукты фторирования ТФЭ в среде фторорганических соединений (нажмите на изображение для загрузки в pdf)
СПИСОК ЛИТЕРАТУРЫ
[1]. Соединения фтора. Синтез и применение. / Под ред. Н. Исикавы. М.: Мир, 1990.
[2]. S. Rozen
// Асc. Chem. Res. 1988. Vol. 21. P. 307.
[3]. Purrington S.T., Kagen B.S., Patric Т.В. // Chem.
Rev. 1988. Vol. 86. P. 997/
[4]. Рахимов А.И. Химия и технология фторорганических соединений.
М.: Химия, 1986.
[5]. Lagow R. J., Margrave J. L. // Progr. Inorg. Chem. 1979. Vol. 26. P.162.
[6]. Florin R. E.// J. Fluor. Chem. 1973. Vol. 14. P.253.
[7]. Hayes L. J., Dixon D. D.
// J. Fluor. Chem. 1977. Vol. 10. P.1.
[8]. Widdecke H. // Proc. 31 IUPAC Macromol. Symp. Merseburg.
1987. Microsymp. 2. 3. S. I. 140.
[9]. Мухаметшин Ф.М . // Успехи химии. 1980. Т. 49. С.1260.
[10]. Scherer K.V., Ono T., Yamanouchi K. Пат. 4626608б (1986). США
[11]. Харитонов А.П.,
Москвин Ю.Л., Колпаков Г.А. Пат. 1754191(1990). Россия
[12]. Halasz S. P., Kluge F., Martini
T. // Chem. Ber. 1973. Vol.106. P. 2950.
[13]. Заболотских В.Ф., Кочанов А.С., Туинов А.В.,
Маркин И.В. // Журнал Органической Химии. 1994. T.30. C.1219.
[14]. Палета О. // Успехи химии.
1971. T. 40. C. 777.
[15]. Паншин Ю.А., Малкевич С.Г., Дунаевская Ц.С. Фторопласты. Л.: Химия,
Ленинградское отделение. 1978. C.24.
[16]. Merritt R.F., Johnson F.A. // J. Org. Chem. 1966.
Vol.31. P.1859.
[17]. Merritt R.F. // J. Am. Chem. Soc. 1967. Vol. 89. P. 609.
[18]. Bormstein
J., Borden M.R., Nunes F.,Tarlin H. // J.Am. Chem. Soc. 1963.Vol.85. P. 1609.
[19]. Shellhamer
D.F.,Conner R.J.,Richardson R.E.,Hesslay V.L. //J.Org.Chem.1984.Vol. 49. P.5015.
[20]. Sherer
K. V., Ono T., Yamamouchi K., Fernandez R. E., Henderson P., Goldwhite H. // J. Amer. Chem. Soc.
1985. Vol. 107. P.718.
[21]. Scherer K.V. // J. Fluor. Chem. 1986. Vol. 33. P.298.
[22].
Аллаяров С.Р., Сумина И.В., Баркалов И.М., Бахмутов Ю.Л., Асамов М.К. // Изв. ВУЗов. Хим. и Химич.
Технол. 1986. Т.32. С.26
[23]. А.Е. Шилов. Химическая кинетика и цепные реакции. М.: Наука, 1966.
[24]. Allayarov S.R., Barkalov I.M., Kim I.P. // J. Fluor. Chem. 1999. Vol.96. P.57.
[25].
Fernandez R.E. Dissertation of degree Doctor of Philosophy (Chemistry). Southern Carolina. 1987.
[26]. Henderson P.V. // Dissertation of degree Doctor of Philosophy (Chemistry). Southern Carolina.
1987.
[27]. Аллаяров С.Р. // Дис. ...докт. хим. наук. М., 1993.
[28]. Аллаяров С.Р. //
Журнал органической химии. 1994. Т.30. С.1147.
[29]. Аллаяров С.Р., Большаков А.И., Баркалов
И.М. // Высокомолек. Соед. 1987. Т.А29. С.364.
[30]. Аллаяров С.Р., Ким И.П., Баркалов И.М.,
Иванова Л.М., Ильин А.Н. // Химия Высоких Энергий. 1993. Т.27. С.22.
[31]. Allayarov S. R.,
Barkalov I. M., Kim I. P. // Mendeleev Comm. 1992. С.139.
[32]. Scherer K.V., Fernandez R.,
Henderson P.B., Krusic P.I. // J. Fluor. Chem.1987. Vol.35. P.167.
[33]. Аллаяров С.Р., Коновалихин
С.В., Гордон Д.А., Чернышева Т.Е., Баркалов И.М. // Химия высоких энергий. 2002. T.36. C.445.
[34]. Аллаяров С.Р., Михайлов А.И. // Известия Академии Наук. Серия химическая. 2001. C.1142.
[35]. Barkalov I.M., Kiryukhin D.P. // Int. Rev. Phys. Chim. 1994. Vol.13. P.337.
Fluorine notes, Номер 5(72) 2010
Fluorine Notes, 2010, 72, 1-2
