Fluorine Notes, 2010, 71, 1-2
Kinetics of radiative polymerization and co-polymerization of fluorinated acrylates and methacrylates
I.P. Kim1, S. R. Allayarov,1, F.N. Baibikov2, D.A.Dixon.3
1) Institute of Problems of Chemical Physics RAS, Chernogolovka, Russia.
2) FSU Russian scientific centre "Applied Chemistry", Perm Affiliate, Perm, Russia.
3)Chemical Department of Alabama University, Tuscaloosa, USA
Abstract:
Polymerization of acrylates (FA) and methacrylates (FMA) with
perfluorinated substituents was initiated by radicals formed by radiolysis in liquid (300
K) and in solid (77 K). Perfluoroinated substituents cause the reduction of kinetic constant
for the polymer chain termination. During FMA polymerization in liquid quasistationary regime
and gel-effect are marked. During FA radiolysis in liquid the rate of polymerization increases
dramatically due to diffusion behavior of chain termination that starts at the very beginning
of the process. When cooled to 77 K the studied monomers turn into a glassy state. During
γ-radiolysis of glassy monomers accumulation of active centers takes place with the radiative-chemical
yield G~3 (radicals per 100 eV of absorbed power), that may initiate post-polymerization
when the samples irradiated at 77 K are heated then to room temperature. Effective post-polymerization
starts within temperature range between devitrification and melting of the monomer. Elongation
of perfluorinated substituents from C2F4 to C8F17 results in higher post-polymerization temperature zone. EPR spectra of growing polymer radicals
formed through the postradiative polymerization are identical to those of monomers with or
without perfluorinated substituents. The formation of cross-linked structures is suppressed
during FMA + FA co-polymerization. Gel - effect is absent in equimole FA/FMA mixtures.
Key words: radiative polymerization, co-polymerization, acrylates and methacrylates with perfluorinated substituents, calorimetry, EPR
Introduction
Perfluorinated organic substances are in considerable use thanks to their high thermal and chemical resistance [1-4]. FA and FMA with perfluorinated groups of various lengths from C1 to C10 are widely employed in the preparation of cross-linked composites for coating of optical fibers. They improve optical, mechanical, adhesive and water-proof behavior of optical fibers [5, 6]. The fiber technology needs pure polymers without ant admixtures (initiators, etc.). In this connection of great interest is the radiative method for polymerization initiation [7]. The process of in-liquid radiative polymerization of acryl and methacryl acid esters was specified in the monograph [7]. Those esters are completely polymerized even by small integral doses of radiation [8, 9]. From the published data [10] it is well known that the methylacrylate (MA) polymerization rate depends on its phase state. MA may convert to glassy or crystal state when cooled to 77 K. Glassy MA when irradiated easily undergoes polymerization [10] changing from glassy state to supercooled liquid, the polymer yield being ~90%. According to [10] irradiated crystal MA does not undergo polymerization.
The kinetics of FA and FMA radiative polymerization was investigated in [11, 12]. The accumulation kinetics for radicals that initiate FA and FMA polymerization process is still not studied. Besides of that the comparison of the basic kinetic characteristics for radiative polymerization and co-polymerization of those fluorinated monomers and their hydrocarbon analogues is necessary in order to describe the impact of elongation of perfluorinated substituents on radical-chain processes with the participation of fluorinated olefins.
The objective of this our study is to investigate both kinetics and mechanism of radiative polymerization and co-polymerization of acryl and methacryl esters of fluorinated alcohols which molecules involve perfluorinated groups.
Experimental Techniques
In this our study we used fluorinated monomers produced by FSUE RSC "Applied Chemistry" (Perm,
Russia):
CH2=C(CH3)COOCH2C2F4H
(FMA-1), CH2=C(CH3)COOCH2C4F8H (FMA-2),
F2C=C(CF3)COOC2H5 (FMA-3), CH2=CHCOOCH2C4F9 (FA-1), CH2=CHCOOCH2C4F8H (FA-2), CH2=CHCOOCH2C6F13 (FA-3), CH2=CHCOOCH2C8F17 (FA-4), and their hydrocarbon
analogs, CH2=CHCOOCH3 (MA), CH2=CHCOOC2H5 (EA), CH2=C(CH3)COOCH3 (MMA), CH2=C(CH3)COOC7H15 (HMA). Their purity was >99.9%.
The radiolysis of monomers by γ- rays 60Co was conducted in vacuum at 77 K in ampoules made of SK-4b glass that does not give EPR signal under γ-radiolysis. The samples radiolysis was carried out with the help of γ-instrument 60Co "Hammatok-1000". Radiation dose rate was 2.78 Gy/s. The dose rate at the point of irradiation was detected with the help of ferro-sulfate dosimeter. EPR spectra were recorded by desk-size radio-spectrometer PS100.X with automatic processing of EPR spectra. EPRWIN software was used for automatic registration. EPRTOOLS software was used in spectra calculation and simulation. Both EPRWIN and EPRTOOLS are products of "Adani" company (Minsk, Belarus).
The polymerization process kinetics was investigated with the help of differential scanning calorimeter [13]. The same method was applied in our study of the monomers phase state. The rate of samples cooling from room temperature to 77 K was 2 K/s.
Results and discussion
1. Post-radiative low-temperature homo-polymerization of FA and FMA in glassy state
(a) Study on the monomer phase transformation.
In order to conduct post-radiative polymerization at low temperature the monomers under study were cooled to 77 K and γ-radiated. After that those irradiated samples were moved into the calorimeter and heated to room temperature. Low-temperature calorimetry makes it possible to control both phase transformation in the monomer matrix and heat effects in the process of post-polymerization [14].
The reagents phase state determines the rate of their meeting in solid state thus reflecting their mobility. Even in the simplest bimolecular reaction the reagents have to meet first of all. Therefore, the study of phase transformations in the process of monomers unfreezing is extremely important in the research of kinetics of low-temperature post-polymerization of monomers. The phase transitions of monomers under study were measured both prior and after their 60Co γ-radiation at 77 K.
Organic substances frozen at 77 K may exist either in crystal or in glassy state [14]. However, among FA studied after cooling to 77 K only FA-4 was in crystal state, while all remaining FA were glassy. The phase transitions that occur during unfreezing of glassy monomers are exemplified by monomer FA-3 (see Fig.1, curve 1).
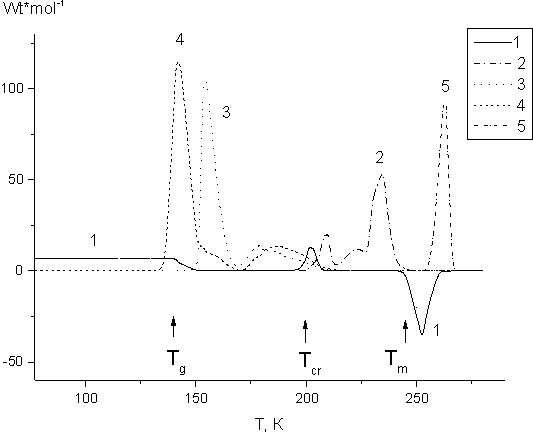
Fig. 1. Calorimetric curves recorded in the course of heating of glassy fluorinated acrylate monomers FA-3 (1,2), FA-1 (3), FA-2 (4) and crystal monomer FA-4 (5) prior (1) and after irradiation of the samples with 10 kGy at 77 K (2-5).
The curve visualizes phase transformations as follows:
(i) glass softens and converts
to supercooled liquid at glass-transition temperature (Tg). The transition is
only followed by the change in heat capacity. During this «glass - supercooled liquid» transition
neither heat absorption nor heat release takes place. It results in a typical step at Tg
on the calorimetric curve for unfreezing;
(ii) supercooled liquid crystallizes
due to the loss in viscosity of the supercooled liquid at Tcr. Its crystallization
is followed by heat release registered as an exothermal peak at Tcr;
(iii)
crystal monomer melts and its endothermic melting peak is registered at the calorimetric
unfreezing curve at Tm.
Glass-transition temperature is generally much below both crystallization and melting temperatures.
Fluorinated acrylate monomer FA-4 being cooled to 77 K forms only crystal state and that is why its calorimetric unfreezing curve contains only melting peak at 265 K.
Calorimetric unfreezing curves for FA irradiated at 77 K are shown in Fig. 1 (curves 2-5). The analysis of those curves proved that post-polymerization of glassy FA occurs in supercooled liquid. Heat release connected with its post-polymerization is only registered in the temperature range Tg -Tcr. Post-polymerization stops during crystallization of irradiated monomers and does not resume after those monomers transition to liquid state. Post-polymerization of FA-3 monomer occurs at the regime of thermokinetic rate oscillation (curve 2, Fig.1). Earlier such regime was observed for post-radiative processes [10] and the mechanism of self-induced oscillations is described in [14] in detail. Decreasing either the heating velocity or the dose for monomer FA-3 radiation one may suppress those thermokinetic oscillations. In the absence of thermokinetic oscillations FA-3 post-polymerization follows equilibrium regime.
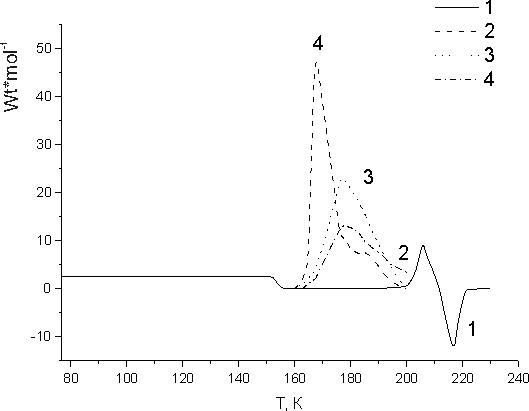
Fig.2. Calorimetric curves recorded during FMA-2 glassy monomer heating prior to irradiation (1) and after its irradiation with doses 10 (2), 25 (3), 110 (4). γ-irradiation was conducted at 77 K.
Calorimetric curve for the post-polymerization of crystal FA-4 is shown in Fig. 1, curve 5. FA-4
post-polymerization is observed within a narrow temperature range of the monomer pre-melting
and melting.
From Fig.1 one may see that elongation of FA perfluorinated substituent
from C4F9 to C8F17 results in higher temperature
zone for its post-polymerization.
Calorimetric studies of fluorinated methacryl monomers FMA-1 and FMA-2 have shown that when cooled
to 77 K they tend to form both crystal and glassy states. It depends on the rate of those
samples cooling. At rapid cooling glassy FMA are formed. Unlike FMA hydrocarbon methacryl
monomers (MMA, HMA) when cooled to 77 K may form only crystal phase [10].
FMA-1 and FMA-2
fluorinated methacrylates do not polymerize under irradiation in crystal state at 77 K, however,
when irradiated in glassy state they undergo effective post-polymerization.
In Fig. 2 calorimetric curves are presented for unfreezing of glassy FMA-2 samples prior to irradiation
(curve 1) and after irradiation with various doses (curves 2-4). One may see that post-polymerization
of glassy FMA occurs similarly to that of glassy FA in viscous supercooled liquid monomers,
when recombination of growing FMA polymer chains is hindered though the monomer rather effectively
diffuse to the reaction centre. At the first step of FMA-2 post-polymerization the monomer
consumption is negligible therefore the dependence of the post-polymerization rate on the
irradiation dose may be deduced from those curves initial regions. Such analysis proved that
FMA-2 post-polymerization rate grows proportionally to the absorbed dose. Besides, those
curves initial regions are easily rectified in Arrhenius coordinates. This made possible
to determine the process general activation energy 71,3±8,5 kJ/mole.
The phase transition
temperatures, specific polymerization heat and melting heat for FA and FMA under study are
present in Table 1.
(b) Investigation of active centers that initiate low-temperature post-polymerization of FMA and FA.
During the irradiation of FMA and FA and their hydrocarbon analogs MA and EA at 77K paramagnetic centers (PC) are being accumulated. The curves corresponding to PC accumulation during the radiolysis of fluorinated monomers (curves 1-6) and their hydrocarbon analogs (curves 7, 8) are present in Fig.3. The analysis of those curves proves that PC accumulation is identical for all studied fluorinated monomers and their hydrocarbon analogs. The radiative-chemical yield of PC calculated from those accumulation curves is G~3. What it means is that about 3 PC are formed per every 100 eV of absorbed energy. The kinetics of PC accumulation under large doses of irradiation was studied by the example of FA-1 fluorinated monomer (curve 1) and MA hydrocarbon monomer (curve 7) irradiated with dose up to 9 MGy (Fig. 3). When the irradiation doses are large the retardation of PC accumulation is only observed starting with irradiation dose = 3 MGy.
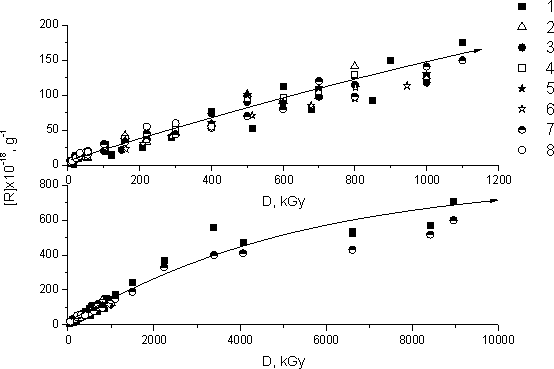
Fig. 3. Graph for the concentration of radicals stabilized during the radiolysis of FA-1 (1); FA-2 (2); FA-3 (3); FA-4 (4); FMA-1(5); FMA-2 (6); MA (7) and EA (8) monomers depending on the dose of irradiation at 77 K.
The EPR spectra analysis has shown that during the radiolysis at 77 K in the matrix of investigated monomers mainly alkyl radicals and, to the lesser extent, vinyl radicals are stabilized [15]. For instance, EPR spectra of FA-3 monomer samples irradiated at 77 K, reveal mainly alkyl radicals CH3-C*HCOOCH2C6F13 and small amount of vinyl radicals CH2=C*COOCH2C6F13. EPR spectra of those radicals are identical to those of similar radicals formed during the radiolysis of acryl acid [16]. The resemblance of EPR spectra was also noted for the radicals stabilized during the radiolysis of FMA-1 and MMA [15].
Heating of monomers irradiated at 77 K is followed by the damage of stabilized active species. The change in PC concentration while heating fluorinated acryl monomer FA-3 (curve 1) and methacryl monomer FMA -2 (curve 2) irradiated at 77 K is shown in Fig. 4.
As it had been proved elsewhere [17] effective recombination of the radicals stabilized in glassy organic matrix occurs usually at temperature ~0.9 Tg. This is due to the unfreezing of translation mobility of the molecule prior to the vitrification of this organic matrix. Analogous temperature dependence of PC damage beginning is observed during FMA-2 and FA -3 samples heating.
On heating of irradiated FMA-2 samples rapid damage of PC begins at ~ 0.9 Tg =140 K (Fig.4, curve 2). Intensive damage of PC continues to FMA-2 crystallization temperature (Tcr =200 K). When FMA-2 is being crystallized the molecule translational mobility is reduced dramatically and about 5% of PC stay isolated in FMA-2 crystal matrix. Slow damage of PC occurs within the temperature range between Tcr =200 K and Tm =215 K. About 3% of all radicals accumulated at 77 K stay stable in liquid FMA-2 after its melting at 215 K. While comparing the curve for unfreezing within the calorimeter (Fig. 2, curve 2-4) with that for the damage of PC (Fig.4, curve 6) for glassy FMA-2 monomer irradiated at 77 K, one may note that effective damage of accumulated PC takes place within FMA-2 Tg -Tcr temperature range, when the monomer post-polymerization is observed.
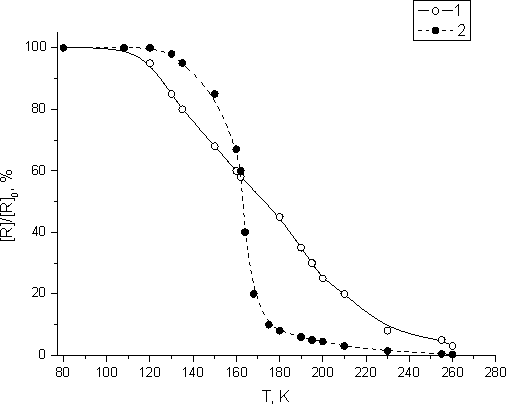
Fig. 4.PC concentration changes under heating of FA-3 (1) and FMA-2 (2) samples irradiated at 77 K, the dose being up to 55 kGy.
The onset of rapid damage during the heating of PC accumulated in irradiated fluorinated acryl monomer FA-3 at 77 K is also observed at temperature ~ 0.9 Tg =126 K (Fig. 3, curve 1). PC damage linearly depends on temperature up to FA-3 melting temperature (Tm =250 K). About 20% of stabilized PC remain stable after FA-3 crystallizing at 200 K. PC isolated in FA-3 crystal matrix are damaged if the sample temperature increases. About 7 % of PC remains stable in liquid after FA-3 monomer melting at 250 K.
From Fig.4 one may see that the curve related to the damage of PC accumulated in FA-3 (curve 1) differs from that of PC temperature dependence in irradiated FMA-2 (curve 2). The rate of PC damage in FA-3 acryl monomer is less than that in of PC damage in FMA-2 methacrylate monomer. However, as in the case of FMA-2 methacryl monomer, in the course of irradiated FA-3 glassy acryl monomer heating post-polymerization occurs within the temperature range between FA-3 devitrification and crystallization (Fig. 1, curve 2), when effective damage of stabilized PC is observed (Fig. 4, curve 1). Those results evidence that the radicals stabilized during those monomers radiolysis at 77 K perform as their initiators of their low-temperature post-polymerization in supercooled liquid.
Deceleration of PC damage in FA-3 acryl monomer is attributable to the growth of its radical’s
thermal stability due to the lengthening of their perfluoroalkyl fragments. FA-3 monomer
involves -C6F13 perfluoroalkyl fragment that is longer than -C4F8H
fragment in FMA-2 monomer. It testifies that long perfluoroalkyl groups have raising impact
on those radicals thermal stability [18]. The effect of alkyl radical’s stability growth
due to the elongation of perfluoroalkyl fragments in organofluoric substances is particularly
notable when it concerns the in-liquid formation of long-lived radicals during radiolysis
[18].
EPR spectra of growing polymer radicals of fluorinated acryl monomer (spectrum
a) and methacryl monomer (spectrum b) are presented in Fig.5. They differ notably. Only
macroradicals with growing polymer chain (~CH2-C*H(COOCH2C6F13)~)
are recorded when intensive post-polymerization takes place during the heating of FA-3 fluorinated
methacryl monomer. EPR spectrum of this radical involves a triplet with splitting ΔH=2.3
mT (Fig.5,a). The validity of the EPR spectrum interpretation is confirmed by the fact that
triplet EPR spectra of "terminal" radicals (~CH2-C*H(COOH)~) with similar parameters
were obtained in the study of polyacryl acid mechanical destruction [19].
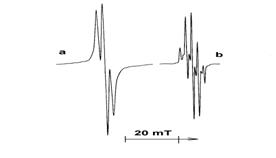
Fig. 5. EPR spectra of FA-3 (a) and FMA-1 (b) irradiated at 77 K with 55 kGy and heated to 150 K. The spectra were recorded at 77 K.
EPR spectrum of irradiated FA-2 methacryl monomer after heating to 150 K reveals the presence
of growing polymer radicals ~CH2C*(CH3)(COOCH2C2F4H)~
(Fig.5, b). Hyperfine structure of the EPR spectrum of such radical comprises nine spectral
lines. It may be interpreted as overlapped quintet and quadruplet with splitting constant
(ΔH=1.5 mT). Growing radicals with similar EPR spectra were recorded in the process of radiative
MMA grafting co-polymerization with polymethylmethacrylate [20].
Those EPR studies
evidence that EPR spectra of growing polymer radicals resulting from postradiative FA and
FMA polymerization are identical to those of growing radicals of their hydrocarbon analogs
without fluorinated substituents.
Table 1. Temperature of phase transitions, specific heat of polymerization and melting for studied monomers
| Monomer |
*Temperatures of phase transitions, K |
Kp*(2Ko)0,5 |
Specific heat, kJ/mole |
||||
|
Tg |
Tcr |
Tm |
Tb |
polymerization |
melting |
||
|
CH2=C(CH3)COOCH2C2F4 (FMA-1) |
160 |
185 |
252 |
420 |
0,28 |
51,4 |
26,5 |
|
CH2=C(CH3)COOCH2C4F8H (FMA-2) |
155 |
200 |
215 |
450 |
0,30 |
39,3 |
10,1 |
|
CH2=CHCOOCH2C4F9 |
130 |
155 |
270 |
75,4 |
3,7 |
||
|
CH2=CHCOOCH2C4F8H |
145 |
195 |
225 |
70,5 |
15,3 |
||
|
CH2=CHCOOCH2C6F13 |
140 |
200 |
250 |
448 |
- |
83,6 |
32,4 |
|
CH2=CHCOOCH2C8F17 |
** |
- |
265 |
98,3 |
24,0 |
||
|
CH2=C(CH3)COOCH3 |
** |
223 |
0,09 |
||||
|
CH2=C(CH3)COOC7H15 |
** |
0,18 |
|||||
*-Tg, Tcr ,Tm , Tb - temperatures of vitrification,
crystallization, melting and boiling;
**- Monomers are not vitrficated and being cooled
to 77 K they form only crystals.
2. Radiative liquid-phase homopolymerization of FA and FMA under γ-radiation at 300 K
The process rates of liquid monomers polymerization at room temperature were measured with the help of a calorimetric setup located directly in the field of 60Co γ - radiation. Thus obtained dependences of the polymerization rates for fluorinated acryl (curve 1) and methacryl (curves 2,3) monomers on the time of irradiation are shown in Fig.6. For comparison with FA polymerization the same dependence for MMA is also shown in Fig.6 (curve 4).
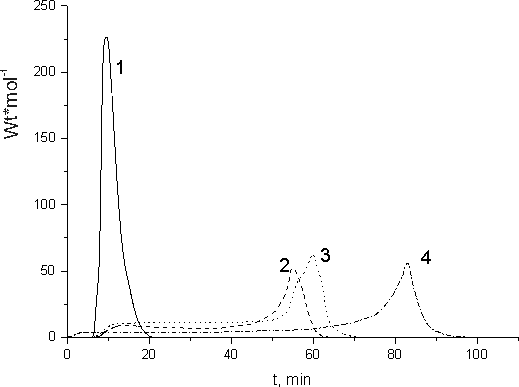
Fig. 6. Dependence of the polymerization rate on the time of irradiation for: 1 - FA-3; 2 - FMA-2; 3 - FMA-1, 4 - MMA. Temperature of irradiation is 313 K.
Induction period due to atmospheric oxygen dissolved in the monomer was only observed for FMA and MMA polymerization. This kind of induction period is practically unavailable during the polymerization of FA-3 acryl monomer. The presence of rather mobile hydrogen in в alpha-position to FA-3 double bond leads to efficient transfer of the chain to the monomer. It results in the formation of strongly branched polymer structure at the most early step of FA-3 radiative polymerization. Quasi-stationary process step is not recorded during radiative polymerization FA-3, and self-accelerated polymerization process was observed immediately after switching of initiative radiation and after short induction period. FA-3 polymerization rate achieves its maximum rapidly and fells down due to the monomer spending. The specificity of this fluorinated acryl monomer liquid-phase polymerization results from the space isolation of its growing macroradicals due to strong branching of their structure at the very beginning of the polymerization chain growth. As a result the probability of their bimolecular damage reduces and their mono-molecular break prevails. The absence of FA-3 polymerization quasi-stationary step was proved by the measurement of the rate of radiative polymerization under the action of γ-radiation of variable intensity.
Therefore, it was shown that the kinetics of FA and FMA radiative polymerization in liquid differ significantly. Similar difference was observed as well during the radiative polymerization of acrylates and methacryl acid esters in [9].
In Fig.7 we show the kinetic curves calculated from the initial sectors of dependencies presented in Fig.6 making use of the monomers polymerization specific heat values. Those polymerization specific heats were determined from the polymer yields and integral polymerization heats. Stationary rate (Wst) exists only at the initial steps of FMA polymerization. The polymerization rate increases dramatically after polymerization of 1-2% of FA-3 or 40-50% of FMA and MMA (Fig.7). The self-accelerated polymerization results from the reduction of the break rate constant (Ko). It is due to the growth of the system viscosity thanks to changeover of the polymerization process to its diffusion regime. Similar acceleration of polymerization earlier was noted during HMA radiative polymerization in [9].
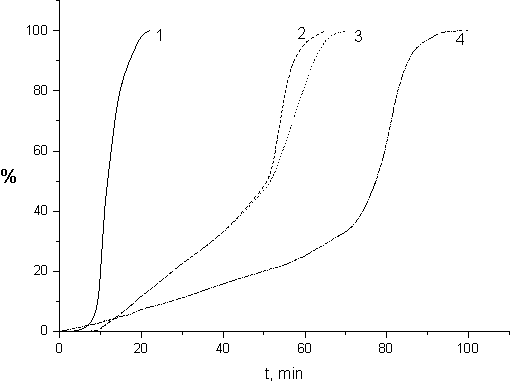 Fig. 7. Dependence of the polymer yield on the time of irradiation calculated from
kinetic curves shown in Fig. 6
Fig. 7. Dependence of the polymer yield on the time of irradiation calculated from
kinetic curves shown in Fig. 6
The kinetic model with quadratic break of polymer chains as depicted in [21] was applied in the analysis of stationary rate of the monomer polymerization. For the studied monomers the following equation was obtained: Wst = Kp* (I/2 Ko)0,5 *[M], here Kp and Ko are the rate constants for the growth and break of growing polymerization chains; I is the rate of radiative initiation; [M] is the monomer concentration.
From the above equation one may see that Wst grows proportionally with I0,5.
To determine the dependence of the polymerization rate on the irradiation dose we studied FMA-2 polymerization at 300 K. The rate of FMA-2 polymerization under variable emitted radiant dose is shown in Fig.8. The analysis of those curves proved that the stationary rate (Wst) is proportional to the 0,5 power of γ-radiation intensity. Therefore, the assumption about quadratic break of growing polymer chains is confirmed experimentally for the case when the degree of polymerization for FMA-2 methacryl monomer does not exceed 40-50 %.
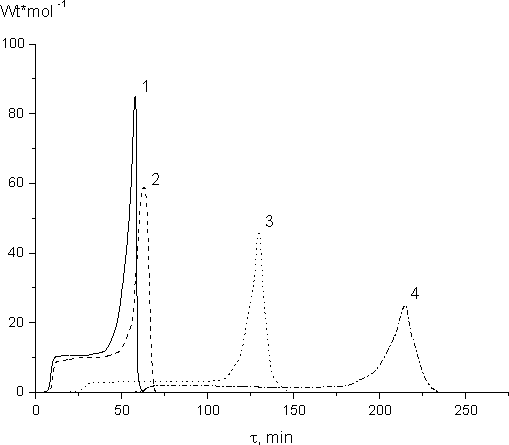 Fig. 8. Dependence of FMA-2 polymerization rate on the time of irradiation at variable
dosage rates, Gy/s: 1 - 5.3, 2 - 2.8; 3 -1.3; 4 -0.6
Fig. 8. Dependence of FMA-2 polymerization rate on the time of irradiation at variable
dosage rates, Gy/s: 1 - 5.3, 2 - 2.8; 3 -1.3; 4 -0.6
We compared the radiative polymerization kinetic parameters for methacryl acid esters with hydrocarbon
(MMA, HMA) substituents and those with perfluorocarbon (FMA-1, FMA-2) substituents. To do
it the values of Kp(2Ko)-0,5 were calculated equal to WstI-0,5[M]-1 on the assumption that absorption of 100 eV of radiation energy results in the formation
of one growing polymer radical. Thus obtained data are present in Table 1. For FMA (FMA-1,
FMA-2) polymerization Kp(2Ko)-0,5 is two times larger than
that for the polymerization of hydrocarbon methacrylates (MMA, HMA). The same reactive methacryl
group is present in all those monomers, therefore their rate constants for chain growth must
be close. The observed increase of Kp(2Ko)-0,5 for FMA is
explicable by the reduction of the rate constant for the break of growing polymerization
chains.
Self-acceleration of MMA polymerization occurs significantly later than in the
case of FMA polymerization (Fig.7). It is due to considerable reduction of the break constant
rate in the case of MMA.
It should be noted that the polymerization of methacryl monomers under study (FMA-1, FMA-2, MMA)
may occur without induction period if the method of for-polymerization is applied (partial
preliminary radiative polymerization).
The impact of different solvents on the kinetics
of acrylates radiative polymerization in liquid was studied by the example of FA-3 polymerization
in the presence of acetone, methane tetrachloride and F2C=CCF3COOC2H5 (FMA-3).
The dependence of polymer yield on the period of FA-3 irradiation in the presence of those solvents is shown in Fig.9. The analysis of this dependence evidences that the presence of up to 30 % by mole of acetone provides no effect on the rate of polymer formation (curve 2). FA-3 radiative polymerization in liquid inn the presence of acetone results in cross-linked insoluble polymer. FA-3 polymerization rate reduces considerably in the presence of (5 - 10) mole % CCl4 (curve 3). When the content of CC14 solvent is 40 - 50 mole % in CC14 + FA-3 blend a totally soluble polymer forms. In that case the process of chain transfer via alpha-hydrogen is completely suppressed and the polymer cross-linking does not occur.
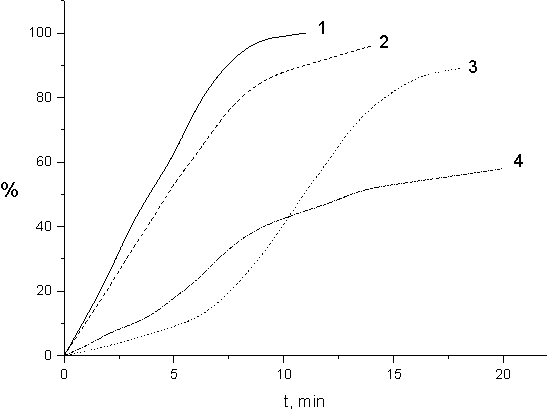
Fig. 9. Dependence of the polymer yield (part by mass) on the time of irradiation (dose) for samples: 1 - FA-3; 2 -FA-3 (70 mole. %) + Acetone (30 mole. %); 3 -FA-3 (91 mole. %)+ CCl4 (9 mole. %); 4 -FA-3 (50 mole. %) +FMA-3 (50 mole. %)
The conducted experiments have shown that FMA-3 monomer (F2C=CCF3COOC2H5) does not undergo polymerization under irradiation neither in liquid (300 K), no in solid state (77 K). That monomer was used as well for solvent in MH-5 polymerization. The dynamics of FA-3 polymerization in FMA-3 medium (Fig. 9, curve 4) differs considerably from that of FA-3 polymerization in mass (Fig.9, curve 1). Initial FA-3 polymerization rate reduces by 5-6 times and its conversion reaches no more than 60% in the presence of FMA-3. The polymer formed in the presence of FMA-3 is dissolved completely in usual solvents. Apparently, the chain transfer via α>-hydrogen does not occur and cross-linked structures are not formed during FA-3 polymerization in the presence of FMA-3. FMA-3 is probably as inert as CC14, and does not participate in FA-3 polymerization.
Specific heats of polymerization shown in Table 1 were determined immediately in the field of γ-radiation at 300 K. Specific heats of polymerization for acryl monomers (FA-1, FA-2,FA-3,and FA-4) are considerably higher than those for methacryl monomers (FMA-1,FMA-2).
From the above data it follows that for the studied monomers their kinetics of low-temperature
post-radiative polymerization differs from that of their radiative polymerization in liquid.
Comparing Fig.1 and Fig.6 one may note that during post-polymerization the initial stationary
section is absent completely, but effective rate of radiative post-polymerization of those
monomers at low temperature (about 180 K) is close to the process rate in liquid (300 K).
Therefore the polymer yield during radiative homo-polymerization of studied monomers
depends on the phase state of irradiated monomer, absorbed irradiation dose, heating velocity
and the solvent nature.
3. Radiative liquid-phase co-polymerization of FA and FMA in the field of γ- radiation at 300 K
The kinetics of FA and FMA co-polymerization was investigated by the example of FA-2 and FMA-2 with the help of low-temperature calorimeter placed directly in the γ- radiation field. The dependence of FA-2 and FMA-2 co-polymerization rate on the time of irradiation for various monomer ratios is presented in Fig.10. The same figure visualizes the rates of FA-2 (curve 1) and FMA-2 (curve 5) homopolymerization for better understanding mutual effects on the polymerization of those co-monomers (curves 2-4). As it was mentioned above (Fig.6) the process of FA homo-polymerization is known to have auto-accelerated regime, and that is why during FA-2 homo-polymerization this self-acceleration is observed immediately after switching of initiating radiation (Fig.10, curve 1). It is caused by effective chain transfer to the monomer that results in the formation of cross-linked structures at the early step of FA-2 homo-polymerization. Homo-polymerization of the fluorinated acryl monomer stops due to the monomer exhaustive expenditure at dose < 2 kGy.
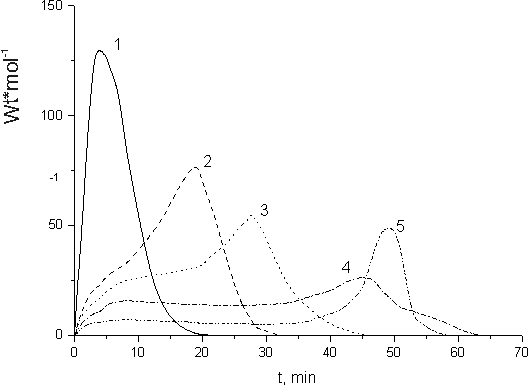
Fig. 10. Dependence of the heat release velocity (polymerization) on the time of irradiation (dose) for: 1 -FA-2; 2 - FA-2 (92.5 mole. %) + FMA-2(7.5 mole. %); 3 - FA-2 (83 mole. %) + FMA-2 (17 mole. %); 4- FA-2 (40 mole. %)+ FMA-2 (60 mole. %); 5 - FMA-2
The observed picture of FMA homo-polymerization differs dramatically when quasi-stationary rate
is set (Fig.4). Quasi-stationary rate of FMA-2 homo-polymerization is established immediately
after switching of γ-radiation (Fig.10, curve 5). Gel-effect is observed only when ~40%
of the monomer is transformed to polymer. Complete conversion of FMA-2 takes place after
its irradiation with dose of 12 kGy. It means that for exhaustive homo-polymerization of
FMA-2 methacrylate requires dose six times larger than that for homo-polymerization of FA-2
acrylate. So different behavior of FA and FMA in radical-chain homo-polymerization reactions
has significant impact on their co-polymerization. The rate of co-polymerization depends
on that monomer to reaction medium ratio. The increase of methacryl monomer (FMA-2) portion
results in the drop of its co-polymerization rate and shifts the start of self-acceleration
(gel-effect) to some later step of the process (Fig.10, curves 2-5). The reaction of FA-2
and FMA-2 co-polymerization causes suppression of the chain-transfer reaction via alpha-hydrogen
of FA-2. This reaction also precludes the formation of cross-linked structures observed in
the case of FA-2 homo-polymerization.
Exhaustive conversion of FA-2 and FMA-2 co-monomers
during their co-polymerization requires less integral dose than that for separate homo-polymerization
of those monomers. Complete conversion of FA-2 and FMA-2 monomers would require 3.4 kGy and
10 kGy, relatively, while in the case of FMA-2 (22 mole. %) + FA-2 (78 mole. %) co-polymerization
the required dose would be 4 kGy.
The co-polymerization behavior pattern for FMA-2/ FA-1 mixture is the same. However, the increase in FMA-2 monomer portion exerts more influence on the rate of FA-1/FMA-2 co-polymerization than on FA-2 / FMA-2. The rate of hydrocarbon monomers EA/MMA co-polymerization depends also on the monomers ratio in reaction media. The reduction of the rate of co-polymerization is observed when MMA content in the initial monomer mixture is built up (Fig.9).
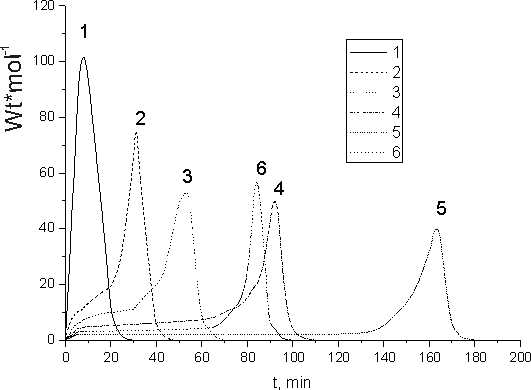
Fig. 11. Dependence of the heat release velocity (polymerization) on the time of irradiation (dose) for: 1 - EA; 2 - EA (95 mole. %) +MMA(5 mole. %); 3 - EA (87.5 mole. %) + MMA (12.5 mole. %); 4 - EA (75 mole. %) + MMA (25 mole. %); 5 - EA (30 mole. %) + MMA (70 mole. %) и 6 - MMA
From the comparison of effect of FMA-2 fluorinated methacrylate on the rate of its co-polymerization with FA-2 fluorinated acrylate (Fig.10) with that of MMA on the rate of its co-polymerization with ЭA (Fig.11) one may see that the maximal retardation of the rate is observed for MMA co-polymerization. Such loss in the rate of co-polymerization has place even in the case of adding (5 - 7) mole. % of MMA.
Exhaustive conversion of monomers during EA + MMA co-polymerization requires higher irradiation dose than their homo-polymerization. For instance, total conversion of EA(30 mole. %)+MMA(70 mole. %) requires 29 kGy, while homo-polymerization of those co-monomers needs only 15 kGy (MMA) and 4 kGy (EA).
Therefore, it is shown experimentally that self-acceleration connected with the formation of cross-linked polyacrylates is suppressed at the co-polymerization of acrylates with methacrylates, both fluorinated and general hydrocarbon monomers. The properties of FA and FMA homo-polymers differ from those of their co-polymers. Usually the properties of FA and FMA co-polymers occupy intermediate position between those of related homo-polymers. Alteration of the initial comonomer composition makes it possible to regulate physical-mechanical properties of resulting co-polymers (elasticity, brittleness, tensile strength) and solubility. E.g., FA homo-polymers form cross-linked structures insoluble by acetone. However, by adding (20 - 30) mole. % of methacrylates (FMA-1, FMA-2) into the reaction medium one may obtain easily soluble FA and FMA co-polymers. It is possible to produce structures with weak cross-linking. It should also be noted that self-acceleration of FA homo-polymerization leads to dramatic temperature and density gradients, that may result in the formation of very inhomogeneous polymers that tend to cracking. Those undesirable consequences may be prevented by the way of FA and FMA co-polymerization. FA and FMA co-polymerization occurs without self-acceleration or sharp temperature gradients.
Co-polymers of acrylates with small amount of methacrylates have more flocculent network than polyacrylates. It is typical both for fluorinated and hydrocarbon monomers. E.g., in acetone the maximal swelling of gel-fraction of co-polymer EA(91 mole. %)+MMA (9 mole. %) is 3200%, while for homo-polymer EA it is only 1500%.
Conducting of FA and FMA radiative co-polymerization allows suppression of the formation
of cross-linked structures at the early steps of the process, and also to level gel-effect
at the later steps of the transformation.
The polymers produced via FMA radiative
low-temperature post-polymerization under small dose of irradiation (2-110 kGy) are easily
soluble in methylethylketone or in acetone. The polymers produced by the radiolysis of
liquid FMA are also easily soluble. Characteristic viscosities of their solutions were
measured at 293 K. They tend to grow with the absorbed dose of preliminary irradiation
at 77 K. E.g., characteristic viscosity values for FMA-2 homo-polymer were 0,6, 0,9 and
1,1 correspondingly when the said dose was 10, 25 and 110 kGy. FMA polymers are very
brittle.
The polymers produced by low-temperature post-polymerization or by polymerization in liquid FA form insoluble net structures. They are sufficiently elastic, and their properties are close to those of caoutchoucs.
CONCLUSIONS
The kinetics of radiative liquid-phase polymerization and co-polymerization of fluorinated
acrylates (FA) and methacrylates (FMA) at 300 K and their low-temperature postradiative
homo-polymerization in glass state was investigated.
The induction period preconditioned
by the air oxygen dissolved in monomer is only observed for FMA and methylmethacrylate
(MMA) polymerization in liquid. This kind of induction period is practically unavailable
during FA polymerization in liquid because strongly branched structures formed at the
early steps of polymerization lead to self-acceleration of the polymerization process.
Therefore it has been found experimentally that FA or its hydrocarbon analogs polymerization
kinetics differs significantly from FMA polymerization by the absence of quasi-stationary
regime. As a consequence the rate grows sharply due to diffusion character of FA growing
chains breakage. The monomer polymerization in liquid is described very well by a kinetic
diagram with quadratic breakage of growing macromolecules and has clearly expressed gel-effect.
The presence of perfluorinated substituents in FA and FMA molecules results in lower
rate constants for the polymer chains breakage.
FMA calorimetric studies revealed that being cooled to 77 K they may form both crystal and glassy state. Among all FA under study only 1,1-dihydro perfluorononyl acrylate forms crystal state when cooled to 77 K, while all other FA convert to glassy state. Glassy FMA and FA polymerize being heated to temperature ranging from devitrification to melting point. The kinetics of FMA postradiative polymerization differs considerably from that of liquid-phase process at 300 K due to the absence of quasi-stationary district. Besides, elongation of perfluorinated substituents from C2F4 to C8F17 results in the elevation of post-polymerization temperature zone by more than 100 K.
During glassy FMA and FA radiolysis at 77 K the accumulation of radicals takes place, their yield being 3 radicals per 100 eV of absorbed energy. The retardation of such paramagnetic centers (PC) accumulation is observed at doses exceeding 3 MGy. The damage of PC in FA occurs slower than in FMA, and the start of rapid PC damage is observed at temperature ~ 0.9 Tg, here Tg is the temperature of vitrification. The radicals stabilized during FA and FMA radiolysis at 77 K initiate their low-temperature post-polymerization. EPR spectra of growing polymer radicals of FA and FMA post-radiative polymerization are identical to those of growing radicals of their hydrocarbon analogs.
Calorimetric study of FA and FMA co-polymerization in the field of γ-radiation was conducted. The presence of FMA (5-10 mole. %) alters dramatically the kinetic parameters of FA radical polymerization. Gel-effect decreases with the growth of FMA content. Considerable addition of FMA (>30 mole. %) suppress formation of cross-linked structure and the co-polymer thus formed is soluble.
LIST OF REFERENCES
2. Panshin Yu.A., Malkevich S.G., Dunaevskaya Ts.S. Ftoroplasty. L.: Khimiya, Leningradskoe otdelenie. 1978. s.24.
3. Karpatova A., Barton J. // Macromol. Chem, 1990, Vol. 191, N 12, p. 2901
4. Kawase K., Hayakawa K. // J. Polim. Sci. Polim. Letters, 1976, vol. 14, N 10, p. 609.
5. Patent US 5690863 (1997)
6. Lekishvili N.G., Nadareishvili L.I., Zaikov G.E., Khananashvili L.M. // Polymers and Polymeric Materials for Fiber and Gradient Optics, 2002, p. 222.
7. Ivanov V.S., Radiatsionnaya khimiya polymerov, Leningrad: Khimiya, 1988, s. 206.
8. Gladyshev G.P., Popov V.A, Radikal’naya polimerizatsiya pri glubokih stepenyah prevrashcheniya. M., Khimiya, 1974, s. 244.
9. Bol'shakov A.I., Kiryuhin D.P., Barkalov I.M. // Vysokomolek. Soed., 1988, t. A30, s. 86.
10.Kim I.P., Kiryuhin D.P., Barkalov I.M. //Vysokomolek. Soed., 1983, t. B24, s. 140.
11.Kim I.P., Barkalov I.M., Bajbikov F. A., Allayarov S.R. // Izv. AN SSSR, 1991, N 6, s. 1330.
12.Kim I.P., Barkalov I.M., Allayarov S.R., Bajbikov F. A. // Khimiya Vysokih Energij, 1992, t. 26, N 2, s. 118.
13.Barkalov I.M., Kiryuhin D.P. // Vysokomolek. Soed., 1980, t. A22, s. 723.
14.Barkalov I.M. //Uspekhi Khimii, 1980, t. 49, N 2, s. 362.
15.Allayarov S.R., Kim I.P., Kispert L.D., // Khimiya Vysokih Energij, in press.
16.Pshezhetskij S.Ya., Tupikov A.G., Milinchuk V.K., Roginskij V.A., Tupikov V.I., EPR Svobodnyh radikalov v radiatsionnoj khimii. M., Khimiya, 1972, 192 s.
17.Ermolaev V.K., Molin Yu.N., Buben N.Ya. // Kinetika i kataliz, 1962, t. 3, s. 58, s. 315.
18.Allayarov S.R. // Zhurnal organicheskoj khimii, 1994, t. 30, N 8, s. 1147/
19.Bresler S.E., Kazbekov E.N., Sominskij E.M. // Vysokomolek. Soed., 1959, t. 1, s. 1374.
20.Milinchuk V.K., Klinshpont E. P., Pshezhetskij S.Ya. // Makroradikaly. M.: Khimiya, 1980, 264 s.
21.Bagdasar'yan H.S. // Teoriya radikal'noj polimerizatsii. 2 izd., M.: nauka, 1966, 300 s.
Fluorine Notes, 2010, 71, 1-2
