Fluorine Notes, 2009, 67, 3-4
IODOFLUOROALKANES: PROPERTIES, SYNTHESIS, APPLICATIONA. V. Morozov, B. N. Maximov Federal State Unitary Enterprise "Russian Scientific Centre (RSC) "Applied Chemistry" The increased interest for iodofluoroalkanes has been created because of their unique properties, which define their value in terms of practice. Since 1967 the studies regarding synthesis of iodofluoroalkanes had been being conducted in RSC of Applied Chemistry. In the present review we have presented the main properties and fields of application for iodofluoroalkanes, the results of studies regarding the development of methods and obtaining technology for iodofluoroalkanes conducted by the authors. 1. Some Physical and Chemical Properties of Iodofluoroalkanes Iodofluoroalkanes - heavy colourless compounds with specific odour and weak narcotic properties. Lowest members of homologous series - gases and liquids, homologues with the number of carbon atoms over 8 - waxes, with the number of atoms over 40 - outwardly remind polytetrafluoroethylene [1]. The density of iodofluoroalkanes is higher than the density of corresponding iodoalkanes, which is because of greater mass of fluorine atoms compare to mass of hydrogen atoms in molecules; it varies within the limits of 2- 2,7 g/cm3. The boiling points of iodofluoroalkanes are on average 60oC lower than the ones of corresponding iodoalkanes through smaller intermolecular interaction caused by the absence of hydrogen bonds typical for iodoalkanes. Iodofluoroalkanes as well as fluorocarbones are characterized by low surface tension, high vapour pressure, small evaporation energies. Iodofluoroalkanes are characterized by strong continuous absorption in short-range UV field. The high value of ionization potentials compare to iodoalkanes (1,25 - 1,4 eV) shows higher delocalization of iodine atoms free electrons through the whole [2]. In consequence of electron-seeking influence of fluorine atoms in the molecule of iodofluoroalkane on the carbon atom connected to iodine a perceptible positive charge is formed, which is partly transferred on easy to polarize iodine atom, and as a result the electron density in the zone of C-J bond is decreasing as well as the value of bond opening energy [3]. Iodofluoroalkanes are not flammable in contrast to iodoalkanes; they dissolve in organic solvents better compare to fluorocarbones. They practically do not dissolve in water. They are moderate toxic compounds. α,ω- diiodoperfluoroalkanes remind monoiodofluoroalkanes in terms of their properties, however their density and boiling points are significantly higher than the ones of corresponding monoiodides. Lowest members of dioiodides series - heavy colourless liquids, the highest ones - waxy compounds [1]. C-J bonds of lowest α,ω- diiodoperfluoroalkanes are less durable than the ones of monoiodofluoroalkanes, but as the CF2J groups are moving away from each other the C-J bond in terms of durability becomes the same as the one of monoiodofluoroalkanes [4]. Both mono- and diiodoperfluoroalkanes turn rose at the light exposition as a result of photo-chemical decomposition with iodide isolation. In terms of their chemical properties iodofluoroalkanes significantly differ from iodoalkanes because of the combination of high inert fluoroalkyl group and reactive iodide in the molecule. It is considered, that C-J bond in iodofluoroalkanes loses its ability for heterolytic decomposition [5]. Because of the partial positive charge in the iodide atom iodofluoroalkanes are not inclined to nucleophilic iodide replacement - the most important type of reaction for [6]; however the nucleophilic substitution is observed at thermal initiation or UV irradiation during reaction with alcohols, ammonia, water with forming of hydrofluoroalkanes [7]. Iodofluoroalkanes are resistant to hydrolysis, but during the boiling together with the alkali aqueous solutions produce hydrofluoroalkanes [8]. Iodine is being substituted by fluorine in iodofluoroalkanes at influence of strong fluorinating agents - F2, CoF3, BrF3, SbF5 [9], medium strong fluorinating agents (JF5, SbF3Cl2) are able to replace iodide for fluorine only in the lowest α, The substitution of iodine for Cl or Br is possible only under the conditions of free-radical reaction [10]. Iodofluoroalkanes are oxidation resistant at routine temperatures, but under the harsh conditions they are oxidized to carboxylic acids under the influence of concentrated H2SO4, HNO3, H2O2, KMnO4 [11]. 2. Iodofluoroalkanes' Application Fields The majority of iodofluoroalkanes' known reactions are free-radical, in which the C-J bond is subject to homolysis during heating, radiation or attack of free radicals. The main field of application for the lowest iodofluoroalkanes - as telogens at telomerization with forming of products used for the synthesis of fluoroacids [11], fluoroalcohols [12], monomers for thermo- and chemically stable polymers [12], at modification of polymers, obtaining of lubricating oils, dielectrics [10] etc. Fluoroalkylation of aliphatic, aromatic, heterocyclic, elementorganic compounds allowing obtaining the compounds of new precisious characteristics [13- 15] is an intensively developing field of application for iodofluoroalkanes. The use of fluoroalkanes in laser systems (optical laser oscillators) is a new direction, which is caused by the ability of iodofluoroalkanes to photodissociation according to C-J bond with forming of high concentration of iodide excited atoms much higher than iodoalkanes can produce [16]. Taking into account the publications on lasers using iodofluoroalkanes and of high power and coefficient of efficiency, this field of their application is becoming important and holds much promise [17]. Lowest monoiodofluoroalkanes find their limited application as highly effective extinguishers for the volumetric fire extinguishing, computer techniques, different types of documentation due to their high thermal stability, high density of vapours, and low toxicity [18]. Due to their insignificant ozone hazard some of iodofluoroalkanes (C2F5J etc.) are used as refrigerants in small refrigerating units and as excellent solvents for cleaning the goods of electronics and precision technics [18, 19]. 3. Main Obtaining Methods For Fluoroalkanes; Advantages and Disadvantages Historically the main obtaining method of iodofluoroalkanes was the replacement of carboxylic group in acids with halogen (iodide), though later iodofluoroalkanes were obtained by the reaction of Hunsdiecker at decarboxylation of fluorocarboxylic acid silver salt with iodide [20, 21]. The chance to obtain iodofluoroalkanes of CF3(CF2)nJ row should be referred to advantages of the method, though the high cost of initial materials, low productivity due to the long period of the process, technological inefficiency, the significant quantity of by-products (10-26% have transferred the method to preparatory class. The method of sodium and potassium salts decarboxylation in the medium of DMF at 150 - 160oC in autoclave [22] is to be known, however the duration of periodical process and significant quantity of by-products (20-30%) bound the introduction of this method. Taking into account the availability and comparatively low cost of the initial raw materials the methods oriented to fluoroolefines appeared to be mostly progressive. The methods of iodofluorination of fluorolefines using the mixture of iodide and JF5 over catalysts got most famous, at that the ratoi of J2 and JF5 was regulated as 2 : 1, which corresponded to the postulated forming at heating up the reagents of JF monofluoride, which produced iodofluoralkane adding by double bond of olefine [24 - 26]. (Ag, Cu, Sn, Sb, Cd, Co, Pb, Ni, Cr) metals [25] were used as catalysts. According the the methods mentioned above the synthesis process was carried out at self developing pressure in autoclaves and at high temperatures (up to 150-225oC). The small productivity caused by the limits of reagents' feeding, long period of the process, instability of the process parameters due to both its periodicity and difficulties of abstraction the heat of exothermal reactions, particularly the telomerization reactions following high heat emission were the methods' disadvantages. The continuous synthesis method of C2F5J directly from J2, F2 and C2F4, is described, in it the gaseous mixture of fluorine and iodine reacts at 150oC forming JF5, which flows down to the column with nozzle, reacting with the mixture of iodine and C2F4 vapours coming from below; the method has not spread because of low yields as well as its potential explosibility [28]. A certain point of view regarding the mechanism of fluoroolefines' iodofluorinating using mixture of iodine and JF5 was not to find in the scientific literature. The forming of JF out of the mixture of iodine and JF5 during the process of iodofluorination postulated by the number of authors did not conform the information of Ruffo and Braida on the absence of JF in such system at room temperature and higher temperatures [29]. Analogously, Schmeisser, Scharf and Sartori [30, 31] stated, that JF as a substance was stable only at low temperatures (under -14oC) and at heating above -14oC dissociated for J2 and JF5. Taking into the account the high temperature of iodofluorination of the mentioned methods [24 - 27], which exceeds the JF temperature of stability, its existence in the processes is unlike to be true. Taking into account the conducted analysis of iodofluoroalkanes synthesis methods described in literature, we developed a method and technology for obtaining of iodofluoroalkanes free from the minuses of known methods, at that the major attention was drawn to iodofluoroalkanes perspective for laser systems using. 4. The Study Of α,ω- Diiodofluoroalkanes Synthesis Process First of all we have studied the thermally initiated reaction of iodine and C2F4 for the purpose of obtaining the 1,2- diiodtetrafluoroethane, out of which it was possible to obtain quantitatively (as we stated) C2F5J, which has got a potential for application in laser systems. Although there are some reactions of iodine and C2F4 initiated photochemically, catalytically or by heating [32 - 34] described in literature, the processes have been stated scarcely, they are characterized by instability and low productivity. We have proved, that gas-cycle reaction of iodine and C2F4 in stationary system passes significantly only above 110oC. Its dependence on inhibitors, on the size of specific area of the reactor, on temperature, which initiated homolysis of molecular iodine into atoms, eased by small amount of energy of J-J bond - 150,7 KJ/mole pointed on radical character of the reaction. The jump in the rate of reaction at increasing the temperature conformed the increasing disintegration of the iodine molecules into atoms which goes with exponential rate constant [16]. S-formed kinetic C2F4 consumption curve at the reaction with iodine looked like the curves of autocatalytic reactions, which was due not to the branching of kinetic chain at the dissociation of formed diiodides into radicals, but to the appearance of the liquid phase out of 1,2-diiodofluoroalkane, during which the reaction was passing at a higher rate. The studies of iodine and C2F4 reaction in the medium of low-polarity solvents, including C2F4J2, allowed proving, that the reaction as a radical chain process was going at as low as 60-100oC; as we can ignore the thermal homolysis of iodine at that temperatures (the equilibrium constant of the J2 →2J reaction is equal to 1,33*10-16 at 100oC), the initiation of radical process, as we had thought was going according to the mechanism of molecularly induced homolysis at donor-acceptor interaction of diluted molecules of iodine and C2F4. The shift of absorption band of the double bond of C2F4 in IR-spectrum (till 1665 cm-1) at introduction of iodine into C2F4,solution tells us about that as well as the appearance of the stretching vibrations band of polarized molecule of J2 at 206 cm-1. Weakened at that interaction J-J bond broke at lower temperature within the range of 60-100oC forming radicals, initiating forming of α,ω-diiodofluoroalkanes. According to the data obtained by our specialists during kinetic studies [36], the rate of interaction of iodine and C2F4 can be described by the equation: r = Keffective.[C2F4]3|2 ∙ [J2]1|2 where Keffectiveis a function of constants of the rates of elementary reactions of initiation, development and break of chains; The discovered regularities allowed developing the obtaining method for 1,2-diiodotetrafluoroethane [35], and also the obtaining method for α,ω-diodoperfluoroalkanes [36]. 5. The Study of Monoiodofluoroalkanes Synthesis Iodofluorination using the mixture of J2-JF5 fluorolefines was studied by our specialists as the most promising direction in the development of iodofluoroalkanes obtaining method. We have chosen the representative of "hard" Lewis acids - SbF3 among accelerating metals fluorides iodo-fluorination process, it provides high rate for the process, as well as it is available and convenient in use. We have proved, that under certain conditions SbF3 in the solution of J2 in JF5 formed complex anions of distorted octahedral symmetry connected by bridge fluorine- SbnF5n+1-, at that the transfer of SbF3 from typical for it p3 condition into Sp3d2 hybrid condition. At acceptor influence of these anions on the easily polarized J2 molecule the molecular cation of J2+ has formed, it enters the composition of the complex of J2+SbnF5n+1-, mainly J2+Sb2F11- (according to the elemental analysis). The analysis of IR- and NMR- spectra of such formations confirmed the structure of the complex [36, 37]: the presence of band at 240 cm-1 in IR-spectrum from molecular cation J2+ , bands at 578 and 692 cm-1 from anions Sb2F11-, bands from bridge bond's vibrations of Sb-F-Sb at 510 cm-1 and (J2+... F-Sb-) at 490 cm-1 bands of degenerate vibrations from four fluorine ligands at each stibium atom; Sb-F-Sb - Yas(SbF4) at 665 cm-1 and YS(SbF4) at 530 cm-1 and 545 cm-1; bands at 620 cm-1 from fluorine in trans position relatively F-Sb-F bridge. The generating of J2+cations in J2 - JF5 iodofluorinating system - the antimony fluorides created the conditions for conducting the synthesis of iodofluoroalkanes at iodofluorinating of fluoroolefines in flow system at moderate temperatures and pressure close to the atmospheric one. At disappearance of J2+cations in the J2-JF5-SbF3 system for example at anhydrous hydrogen fluoride's getting into it, the rate of iodofluorination of fluoroolefine sharply decreased. The study of the J2-JF5-SbF3-HF system using methods of IR- and NMR-spectroscopy revealed the solvation of antimony anions by HF molecules, which hampered their acceptor influence on iodine and formation of J2+ cations. It confirmed the disappearance of the band at 240 cm-1 in IR-spectrum at introduction of HF into iodofluorinating system, the disappearance of the blue tone of solutions, which appeared at the presence of iodine cations in solutions, the alterations of chemical shifts in NMR-F19 spectrum and of signals forms of F19 in the Sb2F11- anion [36, 37]. Solvation of Sb2F11-anions using HF molecules and lowering of catalytic activity of antimony fluorides is reversible: at elimination of HF out of reaction mixture the catalyst restores its activity. The stated connection between the iodofluorinating rate and the rate of fluoroloefines' with J2+cations transformation points out their participation in iodofluorination, and the decreasing of fluorolefines reactivity at iodofluorination in the J2-JF5-SbF3 system, according to the decreasing the electron density of double bonds was the sign of the original electrophilic addition of J2+cation to fluorolefine - the fluorinating rate decreased in the row: C2F4>CF2=CFCl>CF2=CFBr>CF3OCF=CF2>C3F6>C4F8 (as the reactivity towards the electrophiles) The constant of C2F4 iodofluorination rate defined by us significantly exceeded the one during the C3F6 iodofluorination, and the energy of activation was by 8 kJ/mole lower [36]. SbF5 and mixed antimony chorofluorides act at iodofluorination like SbF3, though smaller selectivity and rates of iodofluorination were caused by HF admixture to that associate fluorides, the purification is rather difficult [36]. The interaction of J2+ and fluorolefine begins probably with the formation of π-complex during the equilibrium process, that role of that complex is in the spatial orientation of reagents into the position necessary for reaction. Then the electrophilic atom of iodine cation J2+ forms together with olefin π - electron pair a carbocation or a 3-member iodonium cation. After the addition of F- anion-fluoride and neutralization of charge the iodofluoroalkane forms.
The fact, that isomers form during iodofluorination of fluoroolefines with different substitutes at the double bond ( CF2=CFCl give the mixture of 45% CF3-CFClJ and 55% CF2Cl-CF2J) is the argument for iodonium cation as an intermediate formation. As one side in the iodonium cation is shielded with the iodine bridge, nucleophile F- can attach to any of the two atoms of iodonium cycle carbon from the opposite side. In C3F6 the electrophilic atom of iodine is shifting to the carbon of higher electron density and the orientation of F- anion addition is definite: CF3-CFJ -CF3 is formed. The role of JF5 is in the completing of fluorine ligands of Sb atoms being spent. The synthesis of iodofluoroalkanes in flow system at sparging of fluorolefines through the iodine and SbF3 solution in JF5 was going at creation of saturated solutions of iodine and antimony fluorides; due to the increased rate of reaction the mass exchange processes began to influence the synthesis and could limit the rate of iodofluoroalkanes forming. As the mass transfer of fluoroolefine out of gas bubbles into solution was the limiting stage of synthesis according to our statement [38, 40], the creating of developed interphase surface was necessary for the transferring of synthesis process into kinetic field. It should be noticed, that the synthesis of iodofluoroalkanes under the conditions of barbotage virtually had not been accompanied by the formation of telomers due to the outing of formed products from the reaction zone and low, close to the atmospheric pressure during synthesis. At that, the evaporating heat isolation system provided keeping of isothermal conditions of the synthesis process; taking into account all the heat expenses, the heat effect had not exceeded 0,8 - 7 kJ/mole, while under the static reaction conditions the heating up and decomposition of the mixtures due to the difficulty of heat outing/isolation had been observed [27]. The studies regarding the updating of iodofluoroalkanes synthesis technology in the flow system allowed developing of the mathematical model of C2F4 and C3F6 iodofluorinating processes; the soluability of reagents in JF5 , mass-exchage coefficients, the dependence if interphase surface on the barbotage regime were defined experimentally. Under the simplifying conditions of quasistationary conditions the kinetics and mass-exchange were described. As a result the mathematical model of pentafluoroiodoethane and 2-iodoheptafluoropropane [40] synthesis was composed; under the operational conditions of pilot plant its testing (the model) proved, that the model adequately described the synthesis process of iodofluoroalkanes synthesis. The results obtained during the study of fluorolefines iodofluorination allowed developing of the iodofluoroalkanes obtaining method, which was more up-to-date, than the known ones [39] and as a result the obtaining technologies of pentafluoroiodoethane, 2-iodoheptafluoropropane, trifluoromethoxitetrafluoroiodoethane were developed and introduced at Pilot Plant of RSC "Applied Chemistry". Below you will find the characteristics of obtained iodofluoroalkanes (Table 1). Table 1. Physical Characteristics of Syntheiszed Iodofluoroalkanes
References |
 - diiodofluoroalkanes with forming of monoiodofluoroalkanes [5].
- diiodofluoroalkanes with forming of monoiodofluoroalkanes [5].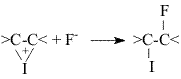
1. Ftor i ego soedineniya. Pod red D.Saimonsa. M. Il.1956, s. 340.
2. Boschi
RA.A. Canad.J.Chem. 1974, V. 52, № 8, p. 1217.
3. Paleta O. Chemike' listy', 1970, r. 64, Č. 3, s. 316.
4. Hauptschein M., Grosse A.V. JACS, 1951,V.73, № 6, p. 2461.
5. Emeléus H.J., Naszeldine R N. J.Chem.Soc, 1949, № 11, p. 2948
6. Nesmeyanov A.N., Nesmeyanov N.A. Nachala organicheskoj khimii; kn.pervaya, M.; Khimiya, 1969, s. 80.
7. Banus J.e.a. J.Chem.Soc. 1950, № 11, p.3041
8. Banus J. e.a. J.Chem.Soc. 1951, № 1, p.60
9. Haszeldine R.N. J. Chem. Soc.1953, № 11, p. 3559.
10. Hauptschein M.e.a. JACS, 1952, V.74, № 3, p. 848
11. Patent US 3351644 (1967).
12. Patent US 3576885 (1971).
13. Tiers G. JACS, 1960, V. 82, № 20, p. 5519
14. Patent US 3271441 (1966).
15. Mc. Joughlin V. Tetrahedron, 1969, V.25, № 24, p. 5921
16. Donohue T. J.Phys.Chem.1976, V 80, № 5, p. 437.
17. Zvelto O. Fizika lazerov-M.; Mir, 1979, s. 197.
18. Patent FR 1443656 (1965).
19. International conference on ozone protection technologies (1996), Washington.
20. Henne A.L., Finnegan W.G. JACS 1949, V 71, № 1. p. 298.
21. Patent US 2554219 (1951).
22. Paskovich D.e.a. J.Org.Chem. 1967, V.32, № 3, p. 833
23. Patent US 3408411 (1968)
24. Patent GB 885007 (1961)
25. Patent US 3006973 (1961)
26. Patent US 3132185 (1964)
27. Patent FR 1385682 (1965)
28. Patent DE 1443534 (1975)
29. Ruffo O. Braida., Z. Anorg. Chem. 1934, B 220. H. 1,S. 43
30. Schmeisser M, Scharf E. Angew. Chem. 1960, ig 72, № 9, p. 324
31. Schmeisser M, Sartori P. Naumann D. Chem. Ber. 1970, Bd. 103, p. 880.
32. Patent JP 11884/68 (1966)
33. Haszeldine R.N. Nature, 1951, V. 167. № 4239, p.139.
34. Coffman D.D. e.a. J.Org.Chem., 1949, 14, p. 747
35. Patent SU 42680 (1967).
36. Morozov A.V. Dissert. k.t.n., L., 1981.
37. Morozov A.V., Maksimov B.N. Tezisy dokladov III Vsesoyuznoj konferentsii po khimii ftororganicheskih soedinenij, Odessa, 1978, s. 68.
38. Patent SU 62465 (1969).
39. Patent RU 2064915 (1993). Morozov A.V., Maksimov B.N. ZH.Or.Kh., 1994, t. 30 v 8, s. 1167
Fluorine Notes, 2009, 67, 3-4
