The Synthesis of Trifluorovinyl Ethers
Yu.V.Zeifman, S.R.Sterlin
The establishment of Russian Academy of Sciences A.N.Nesmeyanov Institute of
Organoelement Compounds RAS Russian Federation,
119991 Moscow, Vaviliva st. 28
Fax: (499) 135 6549,
E-mail: lsg@ineos.ac.ru
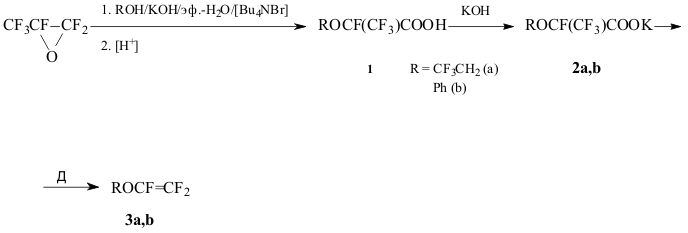
The thermolysis of alkali metals perfluoro-2-alkoxytetrafluoropropionates is
a common method of preparing perfluoroalkylvinyl ethers, including such important
industrial monomers as perfluoromethyl- and propylvinyl ethers, that are
obtained in high yields (<90%)[1]. At the same time in the case of 2-alkoxytetrafluoropropionic
acids 1 the thermal degradation of their salts ROCF(CF3)COOM (2) (R = Me, Et, CF3CH2; M = alkali metal cation)
leads to the formation of complex mixture of products, containing minute
quantities of trifluorovinyl ethers. Thus the thermolysis of 1 (R
= CF3CH2; M = Na) afforded ether 3a in 4% yield [2].
We have shown that ether 3a can be obtained by thermolysis of salt 2 (R = CF3CH2; M = K) (2a) in 67% yield. Similarly
ether 3b was prepared from 2 (R = Ph; M = K) (2b) in
63% yield. The starting acids ROCF(CF3)COOH (1) [R = CF3CH2 (1a); Ph (1b)] were obtained by the reaction of the corresponding
alcohols with hexafluoropropene oxide (HFPO) and alkali in a system H2O-ether
in the presence of phase-transfer catalyst Bu4NBr and subsequent
acidification of the reaction mixture, that simplified the method of synthesis
1 described earlier [3].
The results obtained confirm the conclusion [2] that chemical behavior of salts
2 under the conditions of thermolysis depends to a considerable extent
on the nature of both cation and alkoxy substituent.
2-(2,2,2-Trifluoroethoxy)tetrafluoropropionic acid 1a
Hexafluoropropene
oxide (16,6 g, 0,1 mol) was gradually introduced into the mixture of 19,3g
(0,19 mol) trifluoroethanol, 19g (0,34 mol) KOH, 40 ml of H2O,
50 ml of ether and 1,5g of Bu4NBr at <30oC, then
the mixture was stirred 1h at room temperature, acidified with 30% hydrochloric
acid, ethereal layer was separated, dried with MgSO4 and distilled
to give a fraction with b.p. 82-92oC/35 Torr. The following rectification
afforded 16g (68% based on HFPO taken into reaction) of 1a, b.p. 61-63oC/10
Torr. (lit. data: b.p. 125-127oC [2]). NMR 19F spectrum
(δ, p.p.m.): -3.3 (3F1); 4.0 (3F3);
54,5 (1F2).
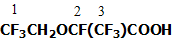
2-Phenoxytetrafluoropropionic acid 1b
was obtained similarly from HFPO and phenol in 40% yield, b.p. 93-98oC/3 Torr; NMR 19F
spectrum (δ,p.p.m.): 5,2 (3F); 42,5 (1F). The acid was transformed
into its K-salt2b without further purification.
2,2,2-Trifluoroethyltrifluorovinyl ether 3a.
A solution of 16g (0,065 mol) 1a in MeOH was neutralized with solution of KOH in MeOH (phenolphthalein
as indicator), evaporated under reduced pressure, the residue was dried over
P2O5 at 110oC/2-3 Torr., then pulverized,
mixed with 20g of dry sand and subjected to thermolysis at 10-15 Torr by
heating in the flame of Bunsen burner (or in Wood alloy bath at 225-280oC).
The volatile products were collected in a trap (-78oC), the condensate
was distilled to give 8.5g (67%) of ether 3a, b.p. 41-44oC
(b.p. and NMR 19F spectrum were identical to that described in
[2,4]).
Phenyltrifluorovinyl ether 3b.
A mixture of 10.4g (0.037 mol) salt
2b (dried over P2O5 at 110-115oC/3
Torr.) and 13g of dry sand was subjected to thermolysis under vacuum of oil
pump, collecting the volatile products in a trap (-78oC). The
condensate was distilled to give 5.9g (63%) of ether 3b, b.p. 132-134oC
(b.p. and NMR 19F spectrum were identical to that described in
[5,6]).
Acknowledgement
Support of the given study by DuPont de Nemours
Co. is gratefully acknowledged.
References
- P.R.Resnick, US Pat. 3692843 (1972)
- M.Y.Pellerite, J.Fluor/Chem., 49 (1990), 43
- D.Sianesi, A.Pasetti, F.Tarii, J.Org.Chem., 31 (1966), 2312
- M.-H. Hang, S.Rozen, UA Pat. 5350497 (1994)
- J.Plummer, L.A.Wall, Trans., 3 (1963), 220
- A.E.Feiring, S.Rozen, E.R.Wonchoba, J.Fluor.Chem., 89 (1998), 31
|
