REACTIVITY OF PERFLUORINATED O-ANIONES
D.P.Del'tsova, V.K.Grinevskaya, L.L.Gervits
A.N.Nesmeyanov Institute of Organoelements
Compounds RAS. 119991, Vavilova str.28, Moscow, Russia
Hydrofluoroethers (HFEs), as a class, are very
promising candidates for substitution ozone-depleting solvents not only
because of their zero ozonedepleting potentials, but also because they
exhibit superior solvent properties. One of the routes for synthesis of
hydrofluoroethers are alkylation of an acid fluoride [1]. Very good
known, that the reversible addition of fluoride-ion to anhydrides of
perfluoronated acids leads to formation of perfluoroalkoxy anions
[2,3,4].
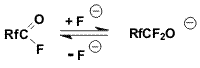
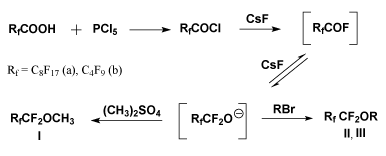
We used both C8F17COOH and C3F7COOH
as the sources of perfluorinated O-anions. The corresponding chlorides
were obtained from these acids by the action of PCl5. For
alkylation of O-anions we used the set of alkylating agents. We assumed
that C8F17CF2O- anion could be prepared
from the corresponding chloride by a one-step procedure with CsF used as
a reagent and with no step of isolation of perfluorononanoyl fluoride.
Actually, we obtained methyl perfluorononyl ether (Ia) in 78%
yield by the reaction of nonanoyl chloride with two moles of CsF in
diglyme followed by addition of dimethylsulfate .
Reactions of O-anions obtained from perfluorononylic and
perfluorobutyric acids with others alkylation agents like allyl bromide,
ethyl bromoacetate and epibromohydrine proceed very slowly with low
conversions and in poor yields.
RBr |
C3F7CF2O- |
C8F17CF2O- |
|
Time of reaction, h |
Yield, % |
Time of reaction, h |
Yield, % |
BrCH2CH=CH2 |
100 |
44 (IIb) |
100 |
28 (IIa) |
BrCH2COOEt |
100 |
18 (IIIb) |
75 |
12 (IIIa) |
 |
15 days
|
No reaction products |
15 days |
No reaction products |
Obviously, equilibrium in the addition reactions of fluoride-ion to
the carbonyl group of an acid fluoride and stabilization of O-anion by
elimination of fluoride-ion is shifted to the left.
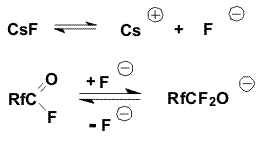
Additionally, the solubility of the corresponding cesium salt in
diglyme is likely to decrease, as the alkyl chain length in the fluoride
increases. Therefore, the rate of alkylation will depend mainly on the
alkylating agent activity. A raise of temperature of the reaction
mixture increases the activity of O-anion, on one hand, but, obviously,
at the same time, additionally shifts the equilibrium to the left, and
the anion concentrations decreases. Under these conditions the
fluoride-ion which presents in the reaction mixture is alkylated to a
greater extent than the O-anion. This is especially pronounced when
halogen-containing compounds are used as alkylating agents. In order to
avoid occurring the reactions of halogen exchange and increase the
activity of an alkylating agents we used a derivative of
p-toluenesulfonic acid, allyl tosylate as the alkylating agents.
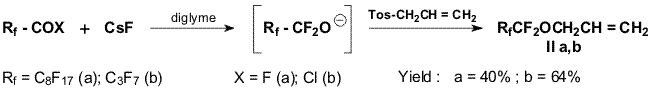
Methylation of perfluorobutoxy-anion with various methylating agents
was carried out at 20oC with the ratio of the starting reagents being
equal.
Table 1
|
Methylating Agent
|
Time, h
|
Yield of Ether (Ib, %) |
|
CF3SO4CH3
|
6 |
70 |
|
CH3SO4CH3
|
15 |
64 |
|
CH3-C4H4-SO3CH3
|
40 |
60 |
|
CH3SO3CH3
|
65 |
51 |
|
CH3I
|
70 |
37 |
Methyl triflate proved to be the most reactive agent. The reaction with
methyl triflate is exothermic and completed for 5 to 6 h. The obtained
ether (Ib) was isolated in 70% yield. The reaction with dimethylsulfate
occurred more slowly, the disappearance of perfluorobutoxy anion (I) was
observed in 12 to 15 h, and the yield of the methyl ether (Ib) was 64%.
Methyl tosylate reacts much more slowly. Reactivity of methyl mesilate
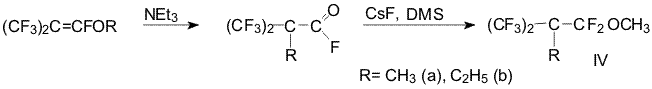
and methyl iodide decrease in series. In order to estimate reactivity
partially fluorinated acyl fluoride we used capability of
perfluoroisobutenyl ethers rearranging to carbonyl fluorides [5]. As we
showered these carbonyl fluorides can add the fluoride-ion and resulted
alcoholates are capable to give the ethers under the action of
alkylation agent with fairly good yield.
It turned out, that trifluoroethylisobutenyl ether don't transform to
corresponding carbonyl fluorides under NEt3 influence. It's interaction
with CsF lead to carbanion, whose methylation affords the corresponding
ether (V).
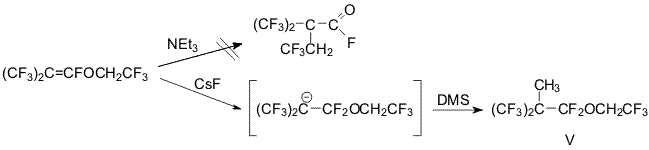
Experimental
1H and 19F NMR spectra were recorded on a
"Bruker AC-200
X" instrument (200 MHz for 1H and 188.3 MHz for 19F NMR, respectively)
with TMS and CF3COOH used as references, respectively. Chemical shifts
are given in ppm using (1H) and (19F) scales, the coupling constants
reported in Hz. Mass-spectra were obtained on a VG 7070E spectrometer
(ionizing electron energy 70 eV).
The course of the reactions was monitored by 19F NMR spectroscopy.
Synthesis of perfluoropelargonyl
chloride
Perfluoropelargonyl chloride (214 g, 85%, b.p. 153 - 155 oC)
was prepared from 241.6 g of perfluoropelargonic acid and 108.6 g of
PCl5 using the procedure reported in J. Am. Chem. Soc. 75, 966 (1958).
Synthesis of methylperfluorononyl ether (Ia)
Chloride from previous
experiment (48.25 g, 0.1 mol) was added slowly dropwise to a suspension
of anhydrous CsF (38 g, 0.25 mol) in 120 mL of dry diglyme. The reaction
mixture was stirred at 20 oC for 16 h, then a solution of DMS (15.12 g,
0.12 mol) in 30 mL of dry diglyme was added, and the mixture was stirred
for 30 h. at 20 oC (the process was monitored by 19F NMR spectroscopy).
The reaction mixture was poured into ice, the lower layer was separated
and dissolved in ether. The ethereal solution was washed with water and
dried over MgSO4. The ether was evaporated, and the residue was
distilled to give ether (Ia) (39.1 g, 78.2%), b.p. 70 - 72 oC /15 mm Hg.
Anal. Calcd for C10H3F19O: C, 24.00; H,0.60; F, 72.20.
Found: C, 24.05;
H, 0.63; F 72.30.
19F NMR ( , ppm): 6.4 t (CF3); 13.8 m (CF2O); 46.4 m
(3CF2); 46.9 m (CF2); 50.2 m (CF2); 51.1 m (CF2);
1H NMR (, ppm): 3.8 s
(CH3). , ppm): 6.4 t (CF3); 13.8 m (CF2O); 46.4 m
(3CF2); 46.9 m (CF2); 50.2 m (CF2); 51.1 m (CF2);
1H NMR (, ppm): 3.8 s
(CH3).
Synthesis of methyl perfluorobutyl ether (Ib) (general
procedure)
Perfluorobutyryl chloride (0.04 mol) was added slowly
dropwise to a suspension of freshly dried CsF (0.1 mol) in dry diglyme
(50 mL) with stirring and ice cooling. The resulting mixture was stirred
for at ca. 20oC, and in 16 h a solution of a methylating agent (0.04
mol) in dry diglyme (15 mL) was added. The reaction mixture was stirred
at ca. 20oC for several hours and volatiles were distilled into a trap
(-78oC) in vacuo (10 mm Hg) at 20-50oC. The content of the trap was
poured into water, the lower layer was separated, washed with water, and
dried over MgSO4. Distillation afforded ether (Ib), b.p. 59-61oC; the
yields and the reaction time are presented in Table 1.
19F NMR (CCl4):
3.7 t (CF3); 11.2 m (CF2O); 48.9 m (CF2); 49.4 m (CF2); J (F-F) = 9.5.
1H NMR (CCl4): 2.98 s (CH3). MS (m/z, assignments, intensity (%)): 249
[M-H]+ 2.24; 232 [M-H2O]+1.32; 231 [M-F]+ 24.82; 219 [M-OCH3]+ 5.15; 197
[C4F7O]+ 2.43; 181 [C4F7]+ 13.04; 170 [C3F7H]+ 1.60; 169 [C3F7]+ 47.51;
151 [C3F6H]+ 1.35; 131 [C3F5]+ 13.41; 119 [C2F5]+ 13.15; 100 [C2F4]+
15.06; 97 [C2F3O]+ 5.17; 93 [C3F3]+ 1.71; 82 [C2F3H]+ 2.62; 81
[CF2OCH3]+ 100; 69 [CF3]+ 60.95; 50 [CF2]+ 2.19; 47 [CFO]+ 10.89; 31
[CF]+ 6.00; 29 [CHO]+ 6.37; 15 [CH3]+ 73.31.
Synthesis of allylperfluorobutyl ether (IIb)
Perfluorobutyroyl
chloride (7 g, 0.03 mol) was added slowly dropwise to a suspension of
anhydrous CsF (11.4 g, 0.075 mol) in 40 mL of dry diglyme. The reaction
mixture was stirred at 20 oC for 16 h, then a solution of allylbromide
(3.6 g, 0.03 mol) in 10 ml of dry diglyme was added, and the mixture was
stirred for 100 h at 50 oC. The reaction mixture was poured into ice,
the lower layer was separated and dissolved in ether. The ethereal
solution was washed with water and dried over MgSO4. The ether was
evaporated, and the residue was distilled to give methyl perfluorobutyl
ether (IIb) (3,7 g, 44,6%), b.p. 92 - 94 oC. 19F NMR (, ppm: -5.9 (CF3)
t J(CF3-CF2)=10 Hz; 10 m (CF2O); 50.5 m (CF2); 50.9 m (CF2) 1H NMR (, ppm):: ABCX2-system: 5.0
ppm. (-OCH2) d. J (CH2-CH )= 5.5 Hz; 5.8 ppm
(=CHB) d. J (HB- CH) = 10.0 Hz; 5.91 (=CHA) d. J (HA- CH) = 17.0 Hz; 6.4
ppm (CH) ddt. J (CH - CHA) = 17.0 Hz, J (CH - CHB)= 10.0 Hz,
J(CH-CH2)=5.5 Hz. MS (m/z, intensity, %) : 276 (M+ C7H5F9O)(16.2).
Synthesis of allylperfluorononyl ether(IIa)
1. Perfluorononanoil
fluoride (30g, 0.06 mol) was added slowly dropwise to a suspension of
freshly dried CsF (15,2 g, 0.1 mol) in 120 mL of dry diglyme. The
reaction mixture was stirred at 20 oC for 16 h, then a solution of allyl
bromide (9,3 g, 0.08 mol) in 30 mL of dry diglyme was added, and the
mixture was stirred at 60 - 70 oC for 100 h. The reaction mixture was
poured into ice, the lower layer was separated and dissolved in ether.
The etherial layer was washed with water and dried over MgSO4. The ether
was distilled off, and distillation of the residue afforded ether (IIa)
(9,7 g, 28,6 %), b.p..80 - 82 oC /10 mm Hg.
2. Using the procedure of
previous experiment the same ether (IIa) was produced (20,1 g, 44%) from
perfluorononanoyl fluoride (39,6 g, 0,084 mol) CsF (15,2 g, 0,1 mol) and
allyl tosylate (18 g, 0,084 mol) in the 150 mL diglyme (50oC, 50 h.) 19F
NMR (, ppm):6.1 (CF3) t J(CF3-CF2)=10 Hz; 10.1 m (CF2O); 46.1 m (CF2);
47.1 m (CF2); 49.7 m (CF2); 50.8 m(CF2). 1H NMR (, ppm): ABCX2-system:
4.7 (-OCH2) d. J (CH2-CH )= 5.5 Hz; 5.46 (=CHB) d. J (HB- CH) = 10.0 Hz;
5.6 (=CHA) d. J (HA- CH) = 17.0 Hz; 6.4 (CH) ddt. J (CH - CHA) = 17.0
Hz, J (CH - CHB)= 10.0 Hz, J(CH-CH2)=5.5 Hz. MS (m/z, intensity, %) :
526 [M]+(19.9).
Synthesis of carboethoxymethylperfluorononyl ether (IIIa)
Perfluoropelargonyl fluoride (4.7 g, 0.01 mol) was added dropwise to a
suspension of freshly dried CsF (2.3 g, 0.015 mol) in 25 ml of dry
diglyme, and the mixture was stirred at 20oC for 16 h. Then a solution
of 1.67 g (0.01 mol) of ethylbromoacetate in 15 ml of dry diglyme was
added. The reaction mixture was stirred at 20oC for 15 h. (According to
19F NMR spectra a minor amount of the alkylation product and
ethylfluoroacetate appeared in the reaction mixture). Then the reaction
mixture was stirred at 50-55oC for 60 h and poured into ice water; the
lower layer was separated and dissolved in ether. The ether solution was
washed with a solution of NaHCO3 , and dried over MgSO4. The ether was
removed, and the residue was distilled to yield ether (IIIa) (0.7 g,
12%), b.p. 110-112oC/4 mm Hg.
1H NMR (CCl4): 1.54 t (CH3); 4.48q (CH2);
4.85 s (CH2); J(H-H) = 7.6 Hz. 19F NMR (CCl4): 5.0 t (CF3); J(F-F)=9.9
Hz; 9.8 m (CF2); -4 to 45.8 m (4CF2); 46.5 m (CF2); 49.1 m (CF2); 50.3 m
(CF2). MS : 572 [M]+ 0.8; 543 [M-C2H5]+ 0.9; 529 [M-C2H3O]+ 0.8; 525
[M-F-CO]+ 3.2; 500 [M- COOC2H4]+ 1.6; 499[M-COOC2H5]+ 15.3; 481
[M-F-COOC2H4]+ 1.5; 480 [M-F-COO22H5]+ 1.9; 219 [C4F9]+6.1;
181 [C4F7]+
2; 169 [C3F7]+ 7.5; 131[C3F5]+ 9.5; 125 [M-C9F17O]+4.4; 119[C2F5]+5.1;
100 [C2F4]+ 4.1; 87[CH2COOC2H5]+ 7.6; 69 [CF3]+ 22; 59 [C3H7]+ 18.6; 45
[C2H5O]+ 3.1; 43[C3H3O]+ 14.6; 31 [CF]+; 29 [C2H5]+ 16.0; 27 [C2H3]+
Synthesis of carboethoxymethylperfluorobutyl ether (IIIb)
Perfluorobutyroyl chloride (4.65 g, 0.02 mol) was added dropwise to a
suspension of freshly dried CsF (6.8 g, 0.045 mol) in 25 ml of dry
diglyme, and the mixture was stirred at 20oC for 16 h. Then a solution
of 3.34 g (0.02 mol) of ethylbromoacetate in 5 ml of dry diglyme was
added. The reaction mixture was stirred at 20oC for 100 h and poured
into ice water; the lower layer was separated and dissolved in ether.
The ether solution was washed with a NaHCO3, water, and dried over
MgSO4. The ether was removed, and the residue was distilled to yield
ether (1.2 g, 18%), b.p. 72-74oC/15 mm Hg. 1H NMR (CCl4): 1.67 t (CH3);
4.61q (CH2); 4.9 s (CH2); J(H-H) = 7.6 Hz. 19F NMR (CCl4): 5.0 t (CF3);
J(F-F)=9.9 Hz; 9 m (CF2); -4.9 m (CF2); 50.8 m (CF2). MS (m/z,
intensity,%, ): 322 [M]+ 0.7; 293 [M-C2F5]+ 2.1; 279 [M-C2H3O]+ 3.5; 275
[M-F-CO]+ 1.1; 250 [M- COOC2H4]+ 2; 249[M-COOC2H5]+ 42; 231
[M-F-COOC2H4]+ 1.15; 230 [M-F-COO2H5]+ 2.4; 219 [C4F9]+ 13.6; 169
[C3F7]+ 2.1; 131[C3F5]+ 8.8; 125 [M-C9F17O]+ 3.2; 119[C2F5]+ 2.5; 100
[C2F4]+ 5; 87 [CH2COOC2H5]+ 6.3; 69 [CF3]+ 31.3; 59 [C3H7O]+ 2.3; 45
[C2H5O]+ 3; 43 [C2H3O]+ 10.2; 31 [CF]+8.8; 29 [C2H5]+ 100; 27 [C2H3]+
Synthesis of methyl 2-methylperfluoroisobutyl ether(IVa)
To a
suspension of freshly calcined CsF (21,5g; 0,14 mol) in 40 ml of dry
diglyme α-methyl perfluoroisobutiryl fluoride (15g, 0,07 mol) was added
dropwise and stirred at ca. 20oC. After 1 h. a solution of DMS (8,9g;
0,07 mol) in 10 ml of dry diglyme was added dropwise and reaction
mixture was stirred for 10 h. at 20oC. The reaction mixture was poured
into ice, the lower layer was separated and washed with water and dried
over MgSO4. Distillation afforded ether (IVa) (9,6g, 55%) B.p. 91°C.
Anal. Calcd. For C6H6F8O: C 29,26; H 2,43; F 61,78%.
Found: C 29,22; H
2,43; F 61,69%.
1H NMR: 1,29 s(CH3); 3,38 s (OCH3) 19F NMR: -7,2
t[(CF3)2]; 3h(OCF2); J F-F = 10,9 Hz.
Synthesis of methyl
2-ethylperfluoroisobutyl ether (IVb)
Using the procedure of previous
experiment methyl 2-ethylperfluoroisobutyl ether was prepared from
?-ethylperfluoroisobutyryl fluoride ( 11,3g, 0,05 mole), CsF (15,2g, 0,1
mole) and DMS ( 6,3g, 0,05 mole). Reaction mixture was stirred for 40 h.
at 20oC and additionally 10 h at 70oC. Distillation afforded ether (IVb)
(4,1g; 31%) B.p. 113-114oC.
Anal. Calcd for C7H8F8O; C 32,30; H 3,07; F
58,48%.
Found: C 32,10; H 2,96; F 58,40%.
1H NMR: 1,44 t(CH3), 2,35
q(CH2), 3,86 s (OCH3); J H-H= 7,2 Hz
19F NMR: -10,2 t(CF3), 0,19
h(OCF2), J F-F=10,8 Hz.
Synthesis of trifluoroethyl
2-methylperfluoroisobutyl ether (V)
To a suspension of freshly calcined
CsF ( 45g,03 mole) in 80 ml of dry diglyme trifluoroethyl
perfluoroisobytenyl ether ( 56g, 0,2 mole) was added dropwise and
stirred at 20oC. After 1 h a solution of DMS ( 25,2g; 0,2mole) in 20 ml
dry diglyme was added dropwise and reaction mixture was stirred at 20oC
for 40h and poured into ice. The lower layer was separated, washed with
water, dried over MgSO4. Distillation afforded trifluoroethyl ether (V)
( 40,9g; 65%) B.p.105-106 oC.
Anal. Calcd. for C7H5F11O: C 26,75; H
1,57; F 66,56%.
Found: C 26,84; H 1,70; F 66,24%. 1H NMR: 1,9 s(CH3);
4,5q(OCH2), JH-F=7,5 Hz. 19F NMR: 1,0h(OCF2); -0,8t(CF3); -7,05
t[(CF3)2C]; J F-F =10,5 Hz; J H-F=7,5 Hz
References
1. D.R. Vitcak, R.M. Flynn. US Pat. No 5750797,
12.04.1998.
2. O. Scherer, H.Millauer. De. Pat No 1301807, 28.08.1969.
3. S. Lecolier, D. Boutte, O. Reboul. Fr. Pat No 2287432, 7.05.1976.
4.
W. Navarrini, M. Galimberti, G. Fontana. EP No 1462434, 29.09.2004.
5. I.L.Knunyants, E.G.Abduganiev, M.V.Urushadze, V.A. Livshits,
Yu.E.Aronov, E.M.Rokhlin. Izv.Ac.Nauk, Ser. Khim., No 1, 110-117,
(1971).
|
