Fluorine Notes, 2007, 55, 3-4
The Synthesis of Low-Fluorinated Compounds Based on Polyfluoroaromatic CompoundsG. G. Furin Siberian Branch of Russian Academy of Sciences Novosibirsk N.N. Vorozhtsov Institute of Organic Chemistry of SB RAS Abstract The analysis of obtaining methods for low-fluorinated derivatives of aromatic row including processes of transformation of polyfluoroaromatic compounds had been carried out: using reducing hydrogenolysis of C-F, C-Cl bonds , electrochemical transformation and protodehalogenation by the influence of zinc, copper, nickel in different media. Here the examples on using that processes with the yield of significant semi-products and materials are given. Table of Contents 1. Introduction 2. Approaches to Synthesis of Partly Fluorinated
Benzene Derivatives 3. Protodehalogenation of Poly-Fluorinated Benzene Derivatives Using Metals Reductive hydrodechlorination of chloroaromatic compounds is commonly held in proton solvents or in their mixtures with aprotonic solvents at medium temperatures (25-100 oC). The methods of conducting such reactions are determined by both used transition metals or their compounds and methods generating transition metals at low oxidation rates. At that we can notice the connection between the process conducting conditions, their selectivity, yields of end products and combination of some factors, among which are the natures of transition metal, reducing agent, solvent and additives of different salts. Due to that, while considering the reagent of reducing system it is necessary to take into account the combination of factors, but not only the solvent itself. 3.1. The Effect of Copper and Its Compounds For the first time the reducing process has been carried out by copper influence on chloropolyfluorobenzenes over water in autoclave at heating up to 300 oC [49]. Thus, pentafluorobenzene was obtained out of pentachlorobenzene with the yield equal to 87 % [49]. The method was also spread onto dichlorotetrafluorobenzenes, 1, 3, 5-trichlorotrifluorobenzene and
The method is universal and the presence of electron-acceptor groups in benzene ring does not hamper the process of hydrogenolysis of C-Cl bond. Thus, 2,6-dichloro-3,5-difluoronitrobenzene selectively reduces over the benzoic acid forming 3,5-difluoronitrobenzene [50].
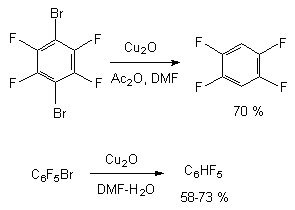
The reduction of brominepolyfluorobenzenes goes much easier. For example, the reduction of bromopentafluorobenzene and 1,4-dibromotetrafluorobenzene using copper oxide (I) over acetic anhydride in dimethylformamide goes during boiling forming pentafluorobenzene and 1,2,4,5-tetrafluorobenzene with the yield of 73% and 70% respectively [51]. For reference the yield of pentafluorobenzene during reduction of bromopentafluorobenzene in dimethoxyethane (85 oC, 24 h) is only 20% [52]. Hydrodebromination of polyfluorobenzenes containing the bromine atom goes under the influence of copper oxide in dimethylformamide solutions, acetic anhydride or in water [51].
Other copper compounds, for example copper chloride (CuCl), are less effective for that process, which can be seen by a low reduction products (4-15 %). The reduction of iodopentafluorobenzene in dimetoxyethane using copper goes very quickly forming pentafluorobenzene (yield equals 98 %) [53]. 3.2. The Action of Zinc in Different Media Using of zinc in proton donor media is a convenient and simple way of hydrodehalogenation of polyfluoroarenes, which goes with high regioselectivity. This to a significant extent is caused by the fact, that zinc has a rather high reductive activity and reacts quite slowly to both subalkali and subacid media [54]. 3.2.1. The Action of Zinc in the Medium of Acids and Bases The boiling of pentafluorobrominebenzene together with zinc in acetic acid produces pentafluorobenzene with the yield equal to 89%, while under the same conditions chloropentafluorobenzene stays the same [55].
In case of hydrodebrominization of dibromotetrafluorobenzenes using zinc in acetic acid the rate of reaction depends on the disposition of bromine atoms - the highest rate can be seen for para-dibromotetrafluorobenzene, at that one or two bonds of C-Br are involved [55].
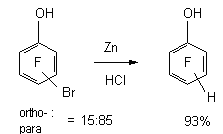
ortho -dibromotetrafluorobenzene, substitution of one Br atom 86%, two Br atoms -14% The mixture of isomeric bromotetrafluorophenoles is reduced by zinc in hydrochloric acid up to tetrafluorophenoles [56].
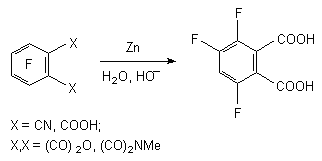
The derivatives of pentafluorobenzoic acid can be reduced by zinc [57-66]. Thus, hydrodefluorination of pentafluorobenzoic acid by zinc in 1 M alkali solution (KOH or NaOH) at 20-100 oC leads to forming of 2,3,5,6-tetrafluorobenzoic acid with the yield of 81.6% [57,67], and pentafluorobenzonitrile produces 2,3,5,6-tetrafluorobenzonitrile in water or aqueous acid with practically quantitive yield [58,59]. The influence of zinc on tetrafluoronitrile in diluted sulphuric acid at 100 oC [60], its influence on tetrafluorophtalic acid, its anhydride [61] or 3,4,5,6-tetrafluoro-N-methylphtalimide [62] in aqueous alkali at 20-160 oC with further hydrolysis in cases of nitrile and imide results in forming of 3,4,6-trifluorophtalic acid with the yield up to 89%.
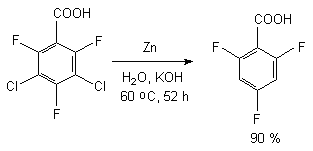
At reduction of 3,5-dichloro-2,4,6-trifluorobenzoic acid using zinc in aqueous KOH at 60 oC during 52 hours we can observe the forming of 2,4,6-trifluorobenzoic acid (concentration in the mixture is 90.5%) [66,68]. Thus we can conduct the selective substitution only of chlorine atoms for hydrocarbon not involving the fluorine atoms of benzene ring.
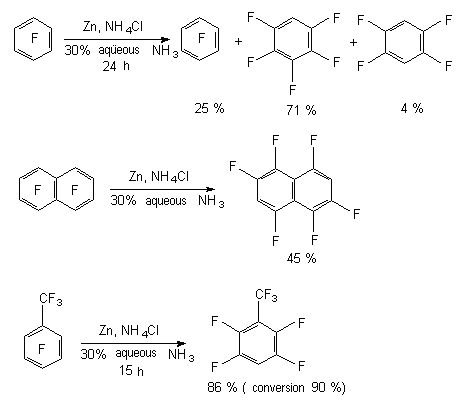
3.2.2. The Influence of Zinc in the Medium of Aqueous Ammonia The most simple modification of reduction system is zinc in aqueous solution of ammonia, which was used as a reagent for a wide range of polyfluorinated derivatives of benzene [69], their functional derivatives [70], polyfluoronaphtalene [69] and pentafluoropyridine [4,71]. Such factors as ammonia concentration, the presence of NH4Cl or organic solvent influence the rate of dehalogenation. At the same time the process doesn't go always with stereoselectivety. Thus, hexafluorobenzene under the influence of zinc in aqueous solution of ammonia over NH4Cl or tetrahydrofurane produces the mixture of partly fluorinated benzenes [69]. The situation like that can happen for octafluoronaphtalene and decafluorodiphenyl. The reduction of octafluoronaphtalene using zinc in aqueous solution of ammonia mainly produces the substitution product for hydrogen of two fluorine atoms out of two rings - 1,2,4,5,6,8-hexafluoronaphtalene. The presence of substituent different from fluorine in benzene ring doesn't hamper the dehalogenation process. For example, octafluorotoluene produces the substitution product of fluorine atom with high yield, which is in para-position towards the CF3 group, for hydrogen atom [69].
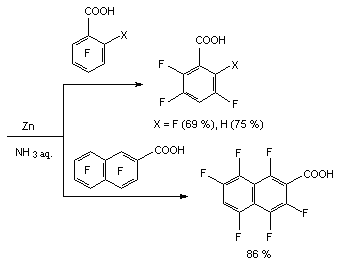
The authors [72] used zinc in 20-30% aqueous solution for reductive defluorinating of perfluorinated carboxylic acids. It was stated, that the process was passing regioselectively and pentafluorobenzoic acid transformed into 2,3,5,6-tetrafluorobenzoic acid, heptafluoro-2-naphtoic acid producing 3,4,5,7,8-hexafluoro-2-naphtoic acid were introduced into it. Hydrodefluorination of 2,3,4,5-tetrafluorobenzoic acid using zinc in aqueous solution of ammonia (20-30%) at room temperature results in forming of 2,3,5-trifluorobenzoic acid [70], and heptafluoro-2-naphtoic acid produces 1,3,4,5,7,8-hexafluoro-2-naphtoic acid in two hours time under these conditions [70].
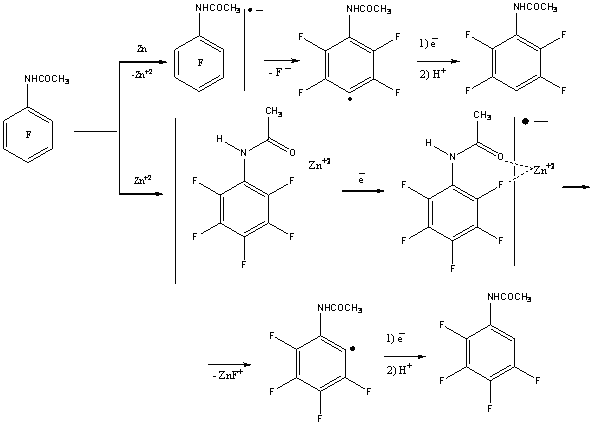
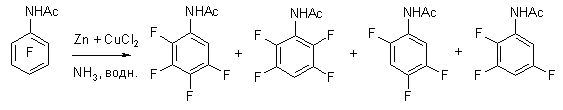
In case of hydrodefluorination of pentafluorobenzoic acid amide using zinc in 20% aqueous ammonia 2,3,5,6-tetrafluorobenzoic acid amide is formed with the yield of 98%. During hydrodefluorination using zinc in aqueous ammonia of N-acetylpentafluoroaniline the authors of work [73] have discovered selective substitution for fluorine hydrogen in ortho-position. They have suggested the following scheme of transformations.
In the work [74] it is stated, that the additive of copper salt increases the rate and depth of pentafluoroacetaniline hydrodefluorination by zinc in aqueous ammonia producing isomeric trifluoroacetanilides.
Analogously hydrodefluorination of perfluoro-4-acetamidediphenyl is carried out.
On a common scale the additive of copper salt allows expanding of application range of zinc-ammonia reducing system for the synthesis of hard-to-reach fluorinated arylamines. Sodium in liquid ammonia can be used as reducing system [72]. For example, 2,3,5,6-tetrafluorobenzoic acid is formed during the interaction of pentafluorbenzoic acid and three equivalents of active zinc in liquid ammonia at -50
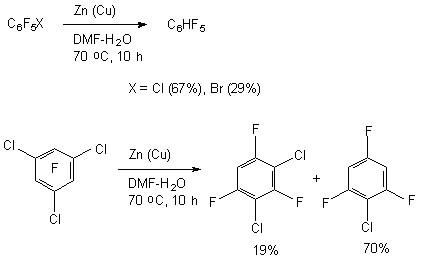
The common preference of C-F bond decomposition in ortho- and para- positions towards metha-position regarding COOH group is observed here [72]. 3.2.3. Hydrogenolysis of C-F Bond on Polyfluoroaromatic Row Under the Influence of Zinc-Copper Pair in Aqueous Dimethylformamide. It has been proved, that in the row of polyfluorochlorobenzenes only C-Cl bonds undergo hydrogenolysis by zinc-copper pair, while the C-F bonds are not involved at that [76-78]. Chloropentafluorobenzene, bromopentafluorobenzene, isomeric dichlorotetrafluorobenzenes and 1,3,5-trichlorotrifluorobenzene at 70 oC easily replace chlorine and bromine atoms under the influence of zinc-copper pair in aqueous dimethylformamide [77].
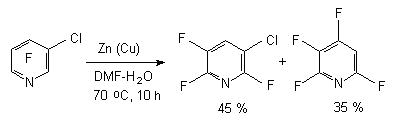
In case of 3-chlorotetrafluoropyridine the influence of zinc-copper pair in aqueous dimethylformamide results in forming of two reduction products - 2,3,5,6-tetrafluoropyridine and 3-chlorine-2,5,6-trifluoropyridine [76,77]. The rate of products depends on the water content in the system. Thus, using the example of fluorochloropyridine we have demonstrated the opportunity of replacement of chlorine atom for hydrogen in position 3 and of fluorine atom in position 4 keeping the chlorine atom in position 3.
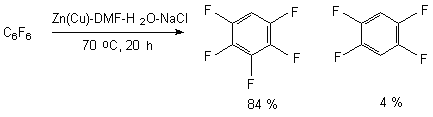
The authors of works [76-78] have demonstrated, that the reductive system of zinc-copper pair in aqueous dimethylformamide is effective for implementing the process of polyfluoroarenes' hydrofluorination. The role of water is both in the accelerating of reductive process and in the influence of reaction direction. The system is a rather high selective one and it can be used for solving the synthetic problems regarding hydrodefluorination and hydrodechklorination of polyfluoroaromatic substrates. It is proved, that its effectiveness depends on the structure of polyfluoroarene being used. Thus, the reductive influence of the Zn(Cu)-DMF-H2O system on hexafluorobenzene goes very slowly (at 70 oC, 10 h the yield of pentafluorobenzene is 1% [27]) and slightly (up to 12 %) will go up, if we add sodium chloride to the system. The effect of electrolytes allows involving a wider range of polyfluoroaromatic compounds into this process.
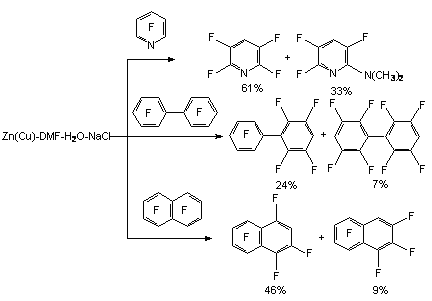
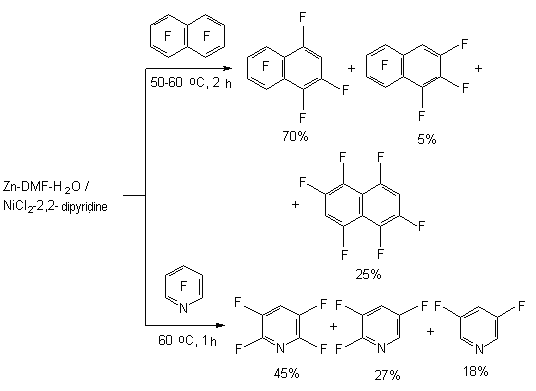
However, only the application of fine-dispersed powders of zinc and copper produces a good yield of target product, which has been demonstrated at a number of examples: pentafluoropyridine, decafluorodiphenyl, octafluoronaphtalene, perfluoroindan and perfluorobenzocyclobutene [76, 77].
Electron-p-type substituents in benzene ring fosters the process of hydrofluorination [76,77,79-82]. In all the cases the fluorine atom is involved, located in para-position towards the substituent [82]. At the same time the introduction of electron-donor substituents, for example, alkoxi-group hampers the reductive process [10,11]. In perfluor-4-tert-butylbenzene the fluorine atoms are replaced in positions 2 and 4, the rate of dimethylformamide and water influences the regioselectivity of the process.
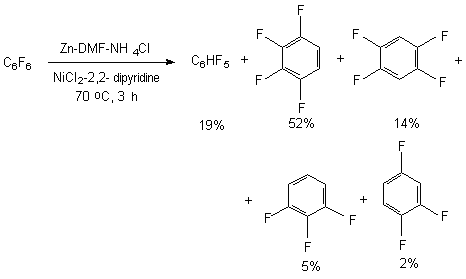
The presence of several electron- p-type substituents leads to selective substitution of fluorine atom in para-position towards substituent as well. It has been demonstrated using the example of hydrodefluorination of perfluoro-ortho-xylene and perfluoro-metha-xylene [77,78]. In case of perfluoro-4-tert-butyltolyol C-F bond undergoes hydrodefluorination, which is situated in ortho-position towards perfluoro-tert-butyl group [83]. These examples demonstrate high selectivity of hydrodefluorination in para-position of benzene ring, and in case of the presence of volume perfluoroalkyl group the substitution for hydrogen of ortho-located . The pattern of hydrogenolysis of C-F bonds in polyfluorinated benzene ring may include the electron's transfer from the surface of zinc towards the polyfluorarene molecule, which leads to the forming of anion-radical. The further decomposition of anion-radical determines the direction of hydrodefluorination reaction [80,84,85]. 3.2.4. Defluorination of Polyfluoroarenes Using Zinc Over The Nickel Salts And Transition Metals Complexes The sharp increase of hydrodefluorinating capacity is observed for the system of zinc-aqueous dimethylformamide in combination with nickel salts. The authors of works [86-88] have proved, that hydrogenolysis of C-F aromatic bond in polyfluoroaromatic compounds is activated by the NiCl2-2,2'-dipyridine and 1,10-phenantrolin-zinc system in the medium of dimethylformamide or dimethylacetamide over the water or ammonium chloride. Along with that a complicated mixture made of fluorobenzenes of different number and fluorine atoms position forms out of hexafluorobenzene.
The presence of zinc increases the conversion of hexafluorobenzene up to 100% (in 4 hours), and the proportion of products amounts to: 45, 11, 18 and 15%. Adding of complexes of titan, zirconium and rhodium to that system results in forming of only 1,2,4,5-tetrafluorobenzene. Octafluoronaphtalene and pentafluoropyridine are introduced into the present process of hydrodefluorination.
It has been found [86,88,89], that polyfluorochloroaromatic compounds are transformed into corresponding polyfluoroarenes under the influence of reductive system generated from zinc and catalytic quantities of NiCl2 and 2,2'-dipyridyl (or 1,10-phenantroline) in aqueous dimethylformamide.
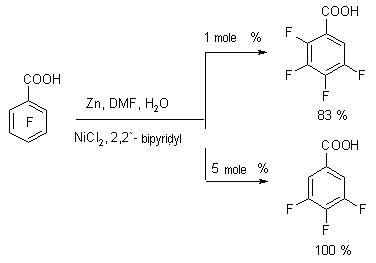
The presence of protons source is necessary for suppressing the reaction of reductive combination with forming of symmetrical diphenyls (optimal rate of DMF: water is 4 : 1, temperature 80 oC). The presence of complex compound of nickel and 2,2-bipyridyl is a decisive factor which determines the reaction pattern. At hydrodehalogenation of polyfluorochlorobenzoic acids using zinc over the metals complexes a selective ortho-position fluorine atom substitution for hydrogen atom takes place, which is used to obtain 2,3,45-tetrafluorobenzoic acid. Thus, at Zn influence on pentafluorobenzoic acid over the NiCl2 (2,2'-dipyridine or 1,10-phenantrolin) in DMF-H2O (or NH4Cl) 2,3,4,5-tetrafluorobenzoic acid of high yield is formed [90]. The depth of transformation depends on the quantity of catalytic system used.
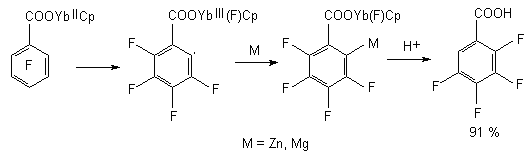
Regioselective defluorination of penatfluorbenzene has been conducted using complex of YbCp2(dme) in tetrahydrofurane over MgI2 at room temperature, the yield of 2,3,4,5- tetrafluorobenzoic acid amounted to 91 % [91,92].
It is supposed, that the process goes through intramolecular transfer of ortho-located fluorine atom into coordination sphere of rare earth element.
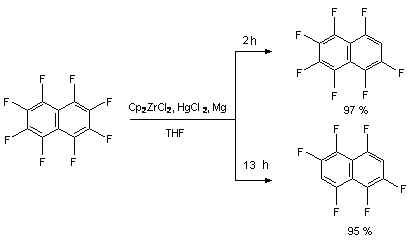
Richmond and his co-workers [93] had demonstrated an opportunity of carrying out the hydrogenolysis of aromatically bound C-F bond by metallocenes of elements of IVB group (Cp2ZrCl2) over mercuric hydrochloride and metallic magnesium at room temperature Thus, they had obtained either mixture of products of 1,3,4,5,6,7,8-heptafluoronaphtalene and 1,3,4,5,7,8-hexafluoronaphtalene [93,94] in two hours or only 1,3,4,5,7,8-hexafluoronaphtalene [93], both products were obtained out of octafluoronaphtalene and showed high yield. Thus, the reaction period is a decisive factor of hydrogenolysis of a number of C-F bonds.
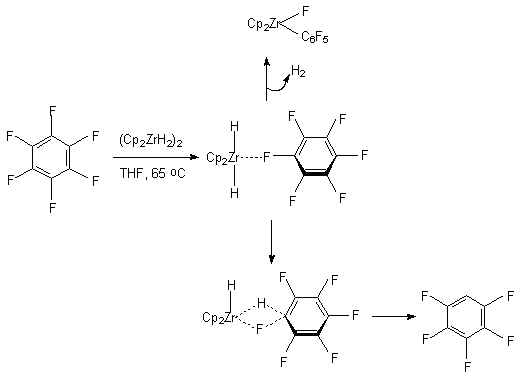
The same system was used for obtaining the pentafluorobenzene (yield was 93%) out of hexafluorobenzene [94]. In case of using the zirconium hydride (Cp2ZrH2)2 the necessity of adding of mercuric dichloride and magnesium dichloride secedes. For example the influence of this compound on hexafluorobenzene in tetrahydrofurane results in forming of pentafluorobenzene [95, 96]. The authors suppose, that the reaction goes with forming of the intermediate compound of Cp2Zr(C6F5)F, in which the C-F bond is activated.
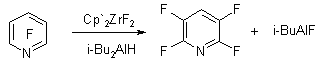
In the work [97] we have demonstrated the opportunity of carrying out the hydrodefluorination of fluorinated pyridines using the influence of Cp'2ZrF2 and diisobutylammoniumhydride i-Bu2AlH.
The (MeC5H4)3U(But) complex was used as well for conducting hexafluorobenzene hydrodefluorination. Thus, the carrying out the hexafluorobenzene and (MeC5H4)3U(But) complex reaction in toluene at room temperature results in forming of pentafluorobenzene as a main product along with C6F5But, iso-butane and isobutene [98,99]. It is proved, that increasing of pentafluorobenzene yield in regard to tert-butylpentafluorobenzene can be reached by increasing the reaction temperature.
Rhodium complexes of [Rh(m-Cl)(COEt)2]2, (Me3P)3RhC6F5 and (Me3P)4RhH over silicon hydrides [Et3SiH, (EtO)3SiH] are able to transform hexafluorobenzene into pentafluorobenzene [10]. It should be noted, that in pentafluorobenzene the C-F bond located at para-position of benzene ring is activated regioselectively, which leads to forming of 1,2,4,5,-tetrafluorobenzene.
References |
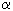
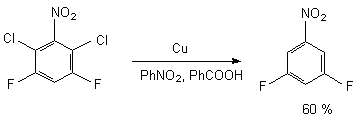
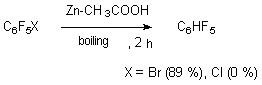

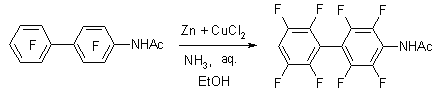
 - 45 oC in 3.5 h, which justifies the preference of defluorination out of para-position compare to other positions. However, as a rule under such circumstances the mixture of benzoic acids with different fluorine atoms is formed. Solvated electron generated by alkali metal in liquid ammonia may be suggested responsible for reducing. Anion-radicals of fluorocontaining benzoic acids were registered by EPR [75]. It should be noted, that really in such system the reduction of the fluorine containing benzoic acid itself doesn't take place, but the ammonium benzoate instead, which forms under the ammonia influencing the acid. Due to that three equivalents of alkali metal are required: one for the reaction involving the ammonia ion and two for the reductive decomposition of C-F bond with further forming of ion fluoride and aryl anion.
- 45 oC in 3.5 h, which justifies the preference of defluorination out of para-position compare to other positions. However, as a rule under such circumstances the mixture of benzoic acids with different fluorine atoms is formed. Solvated electron generated by alkali metal in liquid ammonia may be suggested responsible for reducing. Anion-radicals of fluorocontaining benzoic acids were registered by EPR [75]. It should be noted, that really in such system the reduction of the fluorine containing benzoic acid itself doesn't take place, but the ammonium benzoate instead, which forms under the ammonia influencing the acid. Due to that three equivalents of alkali metal are required: one for the reaction involving the ammonia ion and two for the reductive decomposition of C-F bond with further forming of ion fluoride and aryl anion.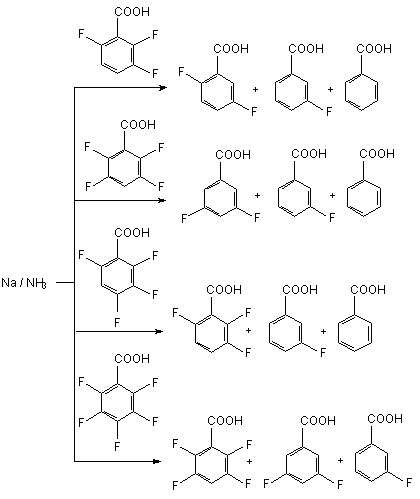
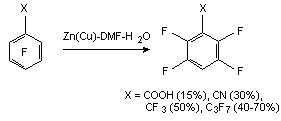
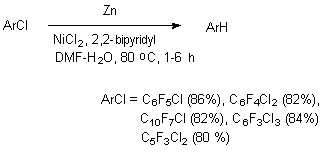
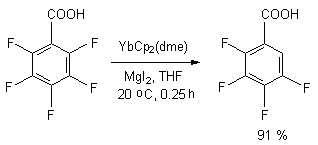
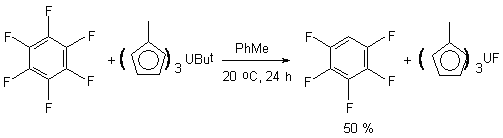

1. Furin G.G., Yumatov V.D. Novoe in Khemie polyfluoroaromatic compounds. Novosibirsk : Pres NGPU (Pedagogichesky Universitet). 2006. 220P.
2. Furin G.G., Fainzilberg A.A. Modern methods for fluorination on organic compounds. M. : Nauka. 239P.
3. Nefedov O.M., Volchkov N.V. Mendeleev Comm. 2006. P. 121-128.
4. Banks R.E., Burgess J.E., Cheng W.M., Haszeldine R.N. J. Chem. Soc., 1965, N 1, p. 575- 581.
5. Hudlicky M. Chemistry of Organic Fluorine Compounds. Ellis Horwood Limited : Chichester. 1976, p. 170-198.
6. Brooke G.M. J. Fluorine Chem., 1997, v. 86, N 1, p. 1-76.
7. Brooke G.M., Burdon J., Stacey M., Tatlow J.C. J. Chem. Soc., 1960, N 4, p. 1768-1771.
8. Holland D.G., Moore G.J., Tamborski C. J. Org. Chem., 1964, v. 29, N 10, p. 3042-3046.
9. Igumnov S., Merkulov K., Igumnova E. 3 International conference “Chemistry, Technology and Application fluorocompounds”. CTAF`2001. 6-9 Juny 2001. St. Petersbusg. Russia. Abstracts. P2-35, p. 221.
10. Kobayashi H., Sonoda T., Takuma K., e. a. J. Fluorine Chem., 1985, v. 27, p. 1.
11. Grady B.J., Dittmer D. J. Fluorine Chem., 1990, v. 50, p. 151.
12. Aizenberg M., Milstein D. J. Am. Chem. Soc., 1995, v. 117, N 33, p. 8674-8675.
13. Brooke G.M., Chambers R.D., Heyes J., Musgrave W.K.R. Proc. Chem. Soc., 1963, p. 213.
14. Schach T., Papenfuhs T. Pat. 5498807 US (1996).
15. Florin R.E., Pummer W.J., Wall L.A. J. Res. NBS, 1959, v. 6, N 3, p. 119-122.
16. Schach T., Papenfuhs T. Pat. 5498807 U.S. (1996); Cl. 507.127; CO 7 25/13).
17. Florin R.E., Pummer W,J., Wall L.A. J. Res. NBS, 1959, v. 6, № 3, p. 119-122).
18. Florin R.E., Pummer W.J., Wall L.A. J. Research Matl. Bur. Standards., 1959, v. 62, p. 119- 122.
19. Rikuo N., Motohiko H. Eur. Pat. 493213 (1992); Chem. Abstr., 199,. v. 117, 233583s.
20. Seisaku K., Takashi S., Hitoshi M. Jpn. Pat. 6310739 (1988); Chem. Abstr., 1988, v. 109, 92449y.
21. Pews R.G., Gall J.A. Pat. 5091580 US (1992); Chem. Abstr., 1992, v. 119, 89943q.
22. Kikuo O., Masami I., Shinichi M., Katsumi T. Jpn. Pat. 04178355 (1992); Chem. Abstr., 1992, v. 117, 233580p.
23. Sshoch T., Papenfuhs T. Eur. Pat. 562435 (1993); Chem. Abstr., 1993, v. 119, 270786x.
24. Osamu K., Nobuo T., Tomoaki N. Jpn. Pat. 6136244 (1986); Chem. Abstr., 1986, v. 105, 78664a.
25. Osamu K., Nobuo T., Tomoaki N. Jpn. Pat. 6122059 (1986); Chem. Abstr., 1987, v. 106, 138094x.
26. Rikuo N., Motohiko H. Jpn. Pat. 04224535 (1992); Chem. Abstr., 1992, v. 117, 251048a.
27. Yakobson G.G., Shteingarts V.D., Vorozhzov N.N. Izv. Akad. Nauk SSSR. Ser. Khim., 1964, № 8, p. 1551.
28. Bardin V.V. Izv. Akad. Nauk SSSR. Ser. Khim., 1997, № 8, p. 1496-1500.
29. Bardin V.V., Pressman L.S. Izv. Akad. Nauk SSSR. Ser. Khim., 1997, c. 819.
30. Bardin V.V., Pressman L.S. Main Group Metal Chem., 1995, v. 18, N 7, p. 333-337.
31. Campbell B.H. Anal. Chem., 1972, v. 44, N 9, p. 1659-1663.
32. Yakobson G.G., Petrov V.P. Izv. SO Akad. Nauk SSSR. Ser. Khim., 1965, Vip. 2, № 7, p. 75-80.
33. Kavin-Miller E., Vajtner Z. J. Org. Chem., 1987, v. 50, N 9, p. 1394-1399.
34. Afanas`ev V.A., Efimov O.N., Nesterenko G.N. et. al. Izv. Akad. Nauk SSSR. Ser. Khim., 1988, № 4, p. 806-809.
35. Efremova N.V., Starichenko V.F., Shteingarts V.D. Izv. Akad. Nauk SSSR. Ser. Khim., 1986, № 12, p. 2794-2796.
36. Efremova N.V., Starichenko V.F., Shteingarts V.D. Zh. Org. Khim.,1988, v. 24, N. 1, p. 57- 68.
37. Efremova N.V., Starichenko V.F., Shteingarts V.D. Zh. Org. Khim., 1992, v. 28, N. 7, p. 1439-1444.
38. Efremova N.V., Starichenko V.F., Shteingarts V.D. Izv. Akad. Nauk SSSR. Ser. Khim., 1988, № 9, p. 2170-2171.
39. Houser K.J., Bartak D.E., Hawley M.D. J. Am. Chem. Soc., 1973, v. 95, N 18, p. 6033- 6040.
40. Starichenko V.F., Shchegoleva L.N., Efremova N.V., e. a. Chem. Phys, 1985, v. 100, N 1, p. 79-87.
41. Starichenko V.F., Selivanova G.A., Shteingarts V.D. Zh. Org. Khim., 1981, v. 17, N. 11, p. 2255-2263.
42. . Selivanova G.A., Starichenko V.F., Ruabinin A.A., Shteingarts V.D. Zh. Org. Khim., 1992, v. 28,Вып. 7, p. 1445-1458.
43. Selivanova G.A., Starichenko V.F., Shteingarts V.D. Izv. Akad. Nauk SSSR. Ser. Khim., 1988, p. 1155; Chem. Abstr., 1989, v. 110, 57208t.
44. Chambers R.D., Musgrave W.K.R., Sargent C.R., Drakesmith F.G. Tetrahedron, 1981, v. 37, p. 591.
45. Chambers R.D., Clark D.T., Sargent C.R. Tetrahedron Lett., 1979, N 21, p. 1917-1920.
46. Drakesmith F.G. J. Chem. Soc., Perkin Trans 1., 1972, N 2, p. 184-189.
47. Yakobson G.G., Platonov V.E., Petrov A.K. Zh. Obshch. Khim. 1966, v. 36. N. 12, p. 2135- 2141.
48. Van der Ham D.M.V., Harrison G.F.S., Spaans A., Van der Meer D. Rec. Trav. Chim., 1975, v. 94, N 7, p. 168-173.
49. Sokolenko V.I., L`vova A.Yu., Nyurin V.S. et. al. Zh. Org. Khim., 1970, v. 6, N. 12, p. 2496- 2498.
50. Kanschik-Conradsen A., Papenfuhs T. Eur. Pat. 557878 (1993); Chem. Abstr., 1994, v. 120, 8325b.
51. Belf L.J., Buxton M.W., Fuller G. J. Chem. Soc., 1965, N 5, p. 3372-3379.
52. Ebert G.W., Rieke R.D. J. Org. Chem., 1988, v.
53, N 19, p. 4482-4488. 53. Ebert G.W., Rieke R.D. J. Org. Chem., 1984, v. 49, N 26, p. 5280-5282.
54. Hudlicky M. Reduction in Organic Chemistry. Ellis Horwood Limited : New York, 1984.
55. Tilney-Bassett J.F. Chem. And Ind., 1965, N 16, p. 693-694.
56. Shtark A.A., Shteingarts V.D. Zh. Org. Khim., 1976, v. 12, N. 7, p. 1499-1508.
57. Nobuo T., Osamu K., Tomoaki N. Jpn. Pat. 60 258143 (1986); Chem. Abstr., 1986, v. 104, 148523j.
58. Masahiko Y., Masanori S., Shusuke N. Jpn. Pat. 01 56656 (1989); Chem. Abstr., 1990, v. 112, 7178d.
59. Masahiko Y., Masanori S., Shusuke N. Jpn. Pat. 02 115156 (1990); Chem. Abstr., 1990, v. 113, 97208m.
60. Masanori S., Masahiko Y., Katsumasa S. Jpn. Pat. 01 160944 (1989); Chem. Abstr., 1990, v. 112, 55243t.
61. Papenfuhs T., Pfirmann R. Eur. Pat. 514863 (1991); Chem. Abstr., 1993, v. 118, 124204u.
62. Fertel L.B., Derwin W.S., Zeffrey S. Eur. Pat. 563986 (1993); Chem. Abstr., 1994, v. 120, 216968r.
63. Masahiko Y., Masanori S., Shusuke N. Jpn. Pat. 01 258639 (1989); Chem. Abstr., 1990, v. 112, 178350h.
64. Masahiko Y., Masanori S., Shusuke N. Jpn. Pat. 02 169542 (1990); Chem. Abstr., 1990, v. 113, 190932c.
65. Masahiko Y., Masanori S., Shusuke N. Jpn. Pat. 02 117643 (1990); Chem. Abstr., 1990, v. 113, 131769g.
66. Osamu K., Nobuo T., Tomoaki N. Jpn. Pat. 6130556 (1986); Chem. Abstr., 1986, v. 105, 114740h.
67. Tominaga N., Kaieda O., Nakamura T. Jpn. Kokai Tokkyo Koho JP 60 258143 (1986); Chem. Abstr., 1986, v. 104, 148523j.
68. Kaieda O., Tominaga N., Nakamura T. Jpn. Kokai Tokkyo Koho JP 6 130556 (1986); Chem. Abstr., 1986, v. 105, 114740h.
69. Laev S.S., Shteingarts V.D. J. Fluorine Chem., 1998, v. 91, N 1, p. 21-23.
70. Laev S.S., Shteingarts V.D. Tetrahedron Lett.., 1997, v. 38, N 21, p. 3765-3768.
71. Selivanova G.A., Chuikov T.V., Shtark A.A., Shteingarts V.D. Zh. Org. Khim., 1988, v. 24, N. 12, p. 2513-2518.
72. Laev S.S., Shteingarts V.D., Bilkis I.I. Tetrahedron Lett., 1995, v. 36, N 26, p. 4655-4658.
73. Shteingarts V.D., Laev S.S., Gurskaya L.Ya., Panteleeva E.V., Selivanova G.A., Beregovaya I.V., Shchegoleva L.N., Vasil`eva N.V. 18th ISFC International Symposium on Fluorine Chemistry. 30th July – 4th August 2006. Bremen. Germany. Anstracts. Lecbure Org. 2.. P. 110.
74. Gurskaya L.Yu., Selivanova G.A., Shteingarts V.D. 7-Vses. Conferenxiy “Chemistry Fluorine@, 5-9 Juny 2006. Moscow. Russia. Abstracts. P-05.
75. Buick A.R., Kemp T.J., Neal G.T., Stone T.J. J. Chem. Soc., 1970, N 13, p. 2227-2231.
76. Krasnov V.I., Platonov V.E., Zh. Org. Khim., 1993, v. 29, N. 5, p. 1078-1079; Chem. Abstr. 1994. Vol. 120. 191201.
77. Krasnov V.I., Platonov V.E. Zh. Org. Khim., 1994, v. 30, N.8, c. 1271-1275; Chem. Abstr. 1995. Vol. 123. 285335.
78. Krasnov V.I., Platonov V.E., Beregovaya I.V., Shchogoleva L.N. Tetrahedron, 1997, v. 53, N 5, p. 1797-1812.
79. Krasnov V.I., Platonov V.E. J. Fluorine Chem., 1991, v. 54, p. 139.
80. Krasnov V.I., Platonov V.E. Izv. Akad. Nauk SSSR. Ser. Khim., 1991, № 9, p. 2158; Chem. Abstr., 1991, v. 115, 279770.
81. Krasnov V.I., Platonov V.E. Zh. Org. Khim., 2000, v. 36, p. 1488; Chem. Abstr., 2001, v. 135, 92398.
82. Krasnov V.I., Platonov V.E. Zh. Org. Khim., 2001, v. 37, p. 517; Chem. Abstr., 2001, v. 135, 357721.
83. Krasnov V.I., Platonov V.E. Zh. Org. Khim., 2001, v. 37, N. 4, p. 552-557.
84. Pierini A.B., Duca A.B., Vera Domingo M.A. J. Chem. Soc., Perkin Trans 2., 1999, N 5, p. 1003-1010.
85. Pierini A.B., Duca A.B. J. Chem. Soc., Perkin Trans 2., 1995, N 7, p. 1821-1828.
86. Adonin N.Yu., Starichenko V.F. Mendeleev Commun, 2000, N 2, p. 60-61: Chem. Abstr., 2000, v. 133, 89412w.
87. Truchin D.V., Adonin N.Yu., Ctarichenko V.F. Zh. Org. Khim., 2000, v. 36, N. 1, p. 143-144.
88. Pat. 2152921 Russia (2000) ; Buleten Isobreteniy. 2000. № 20.
89. Coe P.L., Stephens R., Tatlow J.C. J. Chem. Soc., 1962, N 8, p. 3227-3231.
90. Adonin N.Yu., Starichenko V.F. J. Fluorine Chem., 2000, v. 101, p. 65.
91. Deacon G.B., Pain G.N., Tuong T.D. Inorg. Synth., 1990, v. 28, p. 291.
92. Deacon G.B., Forsyth C.M., Sun J. Tetrahedron Lett., 1994, v. 35, p. 1095.
93. Kipling J., Richmond T.G. J. Chem. Soc., Chem. Commun., 1996, p. 1115.
94. Burdeniuc J., Jedlicka B., Crabtree R.H. Chem. Ber. 1997. v. 130. N 2. p. 145-154/
95. Edelbach B.L., Fazlur Rahman A.K., Lachicotte R.J., Janes W.D. Organometallics, 1999, v. 18, p. 3170.
96. Kraft B.M., Lachicotte R.J., Janes W.D. J. Am. Chem. Soc., 2000, v. 122, p. 8559; 2001, v. 123, p. 10973.
97. Rosenthal U., Jager-Fiedler U., Klahn M., Arndt P., Baumann W., Spannenberg A., Burlakov V. 18th ISFC International Symposium on Fluorine Chemistry. 30th July – 4th August 2006. Bremen. Germany. Anstracts. Inorg 014. P. 330.
98. Weydert M., Andersen R.A., Bergman R.G. J. Am. Chem. Soc., 1993, v. 115, p. 8837.
99. Aizenberg M., Milstein D. J. Am. Chem. Soc., 1995, v. 117, p. 8674.
100. Aizenberg M., Milstein D. Science, 1994, v. 265, p. 359.
Fluorine Notes, 2007, 55, 3-4
