Fluorine Notes, 2004, 36, 3-4
Perfluoroalkylsulphonyl halides, there synthesis and application
E. Bispen, A..Ilyin, D.D.Moldavsky, S. Nurgalieva
(ending)
Lowest alkansulphonylchlorides production doesn't exist in Russia. The production of trifluoromethylsulphonylfluorides using electrochemical fluorination method and with imported raw material is carried out at Angarsk chemical plant. Trifluoromethylsulphonylfluoride is formed with the yield up to 96% [1, 59], but as carbon chain grows the yield of perfluoroalkanesulphonyl halides (PFSH) greatly decreases and fluctuates in the range of 20 - 60% [57 - 59]. Thus, the further analogue C2F5SO2F according to authors data [58] is fluorinated by electrochemical method with the yield up to 60%. At that the reaction mixture contains a great amount of components, close to target products by boiling points, that complicates their isolation.
The additional complicity lies in the fact, that perfluoroethylsulphofluoride mixes with anhydrous hydrogen fluoride (their boiling points are +8,00oC and +9,60oC respectively).
At electrochemical fluorination of aromatic sulphonylchlorides the yield of target products is no more than 4% according to the data [5].
Using of alkylsulphinic acids as raw material also didn't result in success [3, 6]. Besides the low yield (10 - 18%), other additional difficulties arise, connected with formation of water according to the reaction (18)

And this may cause explosion of fluorine oxide, which always forms at interaction of fluorine and water.
Fluorocarbons with the same number of carbon atoms, sulfuryl fluoride (SO2F2) and sulfur hexafluoride (SF6) are by-products of electrochemical fluorination.
The formation of one or another by-product can be explained using one mechanism, but until today there are no sufficient grounds to postulate any definite mechanism of electrochemical fluorination.
As a rule, fluorinated and partly fluorinated products are formed as a result of fragmentation, cyclization, polymerization [5]. Electrochemical fluorination is considered to be an anodic reaction, passing according to free radical mechanism, otherwise the products restored by hydrogen were isolated at cathode [13, 5, 7] The formation of great amount of polymer products, particularly out of aromatic raw material is impossible to combine with ionic mechanism.
In spite of rather high yield of a number of flouroorganic products, the common level of electrochemical fluorination is still far from perfection. Low productivity, a big number of waste products and necessity to purify desired products are the most essential drawbacks of this method. Electrolyzers productivity is not a constant value. At first it increases, then stabilizes, after that decreases due to electrodes corrosion and their screening by resinification reaction products [3, 5, 82, 83].
Attempts of using porous anodes were made. Due to porous surface the electrolyzers productivity increases, but their working period decreases due to more intensive corrosion and resinification.
Taking into account the drawbacks, we can make a conclusion, that for exhaustive fluorination of partly fluorinated alkylsulphonylfluorides this method is not much suitable.
At fluorination of different class organic compounds many authors used metal fluoride method, the essence of which is the substitution of hydrogens for fluorine in hydrocarbons by highest fluorides of transition metals according to the reaction (19)
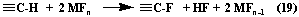
Cerium fluoride (IV), cobalt fluoride (III), silver fluoride (II), nickel fluoride (III), mercury fluoride (II), copper fluoride (II) are considered to be the main fluorinating agents. A lot of works is devoted to sue of the method both in our country [64 - 68], and abroad [60 - 63].
Fluorides of alkali metals and also lowest fluorides of transition metals are used for substitution of functional groups of organic compounds [2 - 6, 27, 69]. The successful using of cobalt trifluoride [60 - 63] forwarded essential widening of nomenclature of fluorooragnic compounds, which were hard to obtain using other methods.
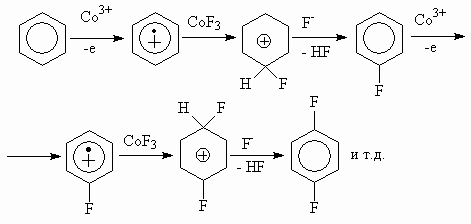
Organic compounds fluorination mechanism using cobalt trifluoride is determined by fluorination conditions and nature of substrate [61, 63, 64]. Thus, for aromatic and unsaturated hydrocarbon cation-radical theory of fluorination [64], which can be illustrated by schemes 20 and 21 was suggested.
| (20) |
For olefines:
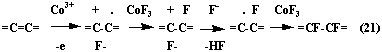
There is no unified mechanism both for cobalt trifluoride and highest fluorides of different metals, because the fluorination mechanism depends on crystal structure of used fluorides and also on thermodynamical stability of radicals and cations [64, 65-1].
At fluorination of ethylene a big amount of asymmetrical isomers of difluoroethane and tetrafluoroethane is formed. It is possible if intermediate ions or radicals regroup according to the scheme (22)
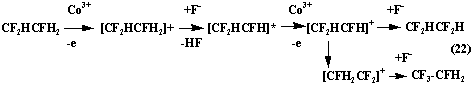
In the works [65, 66] the influence of processes of adsorption-desorption, diffusion, temperature, period of contact and concentration of hydrogen fluoride is studied.
The studying of fluorination kinetics using cobalt trifluoride showed, that a number of substrates can sorb on the surface of CoF3, being for a long time in flow reactor and thereby preventing further fluorination of initial materials [65].
Cobalt difluoride, forming at reaction of hydrocarbons with cobalt trifluoride doesn't inhibit the fluorination process.
The role of hydrogen fluoride is determined by temperature of the process. Thus at temperature which is below 300oC at optimal concentration hydrogen fluoride produces autocatalytic action, speeding up the fluorination process, and accumulation of big quantity of HF on the surface results in stagnation of the process because of diffusive difficulties appearing at transportation of reagents to surface and blocking of reaction centers [65-1].
Thus, rather hard conditions are required to carry out exhaustive fluorination using cobalt trifluoride. The majority of the processes is passing at temperatures 350-450oC with the yield of a number of products ranging from 60 to 83%, the rest is products of thermodestruction, the quantity of which is also determined by the nature of initial materials. Particularly, the contribution of thermodestruction is great at fluorination of compounds, containing different heteroatoms in functional groupings: nitrogen, oxygen, sulfur [64].
The using of this method for exhaustive fluorination of partly fluorinationated compounds (paraffins, ethers, trialkylamines) [66,67,68] provides mostly full fluorination with the yield of a number of products up to 96%.
It is considered, that duration of cobalt trifluoride at its regeneration is unlimited, as researches showed [66-68] cobalt oxides are finally fluorinated to trifluoride at temperatures 500-600oC, fluorinating capacity is restored.
But taking into account the increased cost of cobalt trifluoride at the chemical raw materials market, the production of some compounds using it became unprofitable.
Comparative fluorination using cobalt trifluoride itself and cobalt trifluoride with simultaneous supply of elemental fluorine [68] showed real perspectives of cobalt trifluoride using in production of perfluorinated products (parafines, ethers, amines).
It is stated, that at fluorine using cobalt difluoride is fluorinated to trifluoride at rate higher than rate of cobalt (III) up to cobalt (II) reduction, as a result of which the total fluorination rate stays constant.
The technique of direct fluorination using to obtain perfluorinated compounds is different:
- The using of fluorine dilution by inert gases [1-6]
- using of low temperature reactors with controlled cooling according to zones [74,75,76]
- aerosol method [77], - however, the contribution of destructive fluorination (up to 93%) of C-C bonds (energy dissociation 270 KJ/mol) , resulting from energy isolation in big quantity:
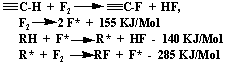
At catalytic fluorination of organic compounds the main function of catalyst is in taking aside of isolated heat [70-73, 78-81]. It allows avoiding local overheating, providing constant temperature gradient in fluorination reactor and it increases selectivity. In connection with this the wide use of metal catalysts, such as cuprum, silver, nickel, possessing higher thermal conductivity is understandable [1-5]. Though in the work it was shown [73], that cuprum, modified by silver is more preferable, than ordinary cuprum. The increasing of selectivity is connected here with catalytic action of silver fluoride AgF2, and role of elemental fluorine is reduced to regeneration of silver fluoride [1].
According to the work [74] the process of direct catalytic fluorination must pass in excess of fluorine; at temperatures higher than boiling points of compound fluorinated, because at lower temperature the initial material will sorb on the surface of catalyst, that inevitably will lead to destruction of carbon skeleton, but lower the catalyst destruction point; direct fluorination requires using of aggressive proof catalysts and reactor materials.
In the works [78-81] results of using such catalysts are listed. They are prepared by soaking of corresponding carrier with salt solvents with their further drying, burning and fluorination. Among most active carriers according to the data [78] is aluminium  -oxide, and most suitable catalyst for hydrogen substitution is nickel difluoride.
-oxide, and most suitable catalyst for hydrogen substitution is nickel difluoride.
Cobalt [70], aluminium [71], antimony and titanium [72] fluorides were used as catalysts of halogen atoms selective substitution for fluorine.
Localization of fluorination acts on the surface of catalyst provides well-timed excessive energy abstraction of activated adducts . At that catalyst, possessing developed surface intensively sorbs fluorine and activates it, that provides kinetic premises of process localization on the surface. The growth of the reaction volume share forwards selective fluorination, that is provided by the developed surface of carrier, and surface concentration of active centers (NiF2) and higher sorption activity for elemental fluorine provides increasing of fluorination speed [67,78].
It was shown, that fluorine containing hydrocarbons can be fluorinated exhaustively using elemental fluorine with high yield (up to 94-97%) [78]. Both low-molecular tetrafluoroethylene production residue and dihydropolyfluorinated compounds with number of atoms ranging from 12 to 15 were used as raw materials. Catalytic fluorination of organic compounds, containing heteroatoms: nitrogen, oxygen, sulfur [79-81] was followed by essential destruction contribution to composition of end products.
Undoubtedly, if we take the energy of bonds formation into consideration :
C-F - 436-507 KJ/mol
C-S - 272 KJ/mol
C-O - 358 KJ/mol
S=O - 746 KJ/mol
C-H - 413 KJ/mol
S-F - 285 KJ/mol
Then high grade of carbon-sulfur bond's destruction becomes clear.
It can be supposed, that it is confirmed by the works of a number of researchers [82,83,92], that
- as hydrogen atoms are being replaced by fluorine, the rate of fluorination decreases, at that not only the heat of fluorination is decreasing, but also the rate of heat-evolution, that makes the use of simple technological equipment possible in principle;
- as carbon chain is becoming longer, the conditions of fluorination become tougher [83]. Thus, polyfluorinated olefins with a number of carbon atoms ranging from 2 to 4 are fluorinated at -40 oC - -100oC, and olefine with a number of carbon atoms starting from 8 are fluorinated at temperature in the range of: 120oC-160oC. It can be explained by increasing of C-F bond energy as fluorine is being accumulated in the molecule of fluorocarbon. [3]
CH3F - 448 KJ
CH2F2 - 459 KJ
CHF3 - 477,5 KJ
CF4 - 507 KJ;
- the higher is the molecule oxidability grade, the more fluorine resistant it is ; dialkyl ethers are more resistant, than paraffin's, olefins and trialklylamines;
- introduction of solvent into the process of direct fluorination lowers the grade of destruction [84-90]
- at fluorination of enamines and unsaturated ethers, at first multiply bonds are being fluorinated [82-84].
At choosing solvent for direct gas-phase fluorination its physo-chemical constants, chemical inertness, the possibility of regeneration are taken into account [85-88].
As a rule, nitrogen and helium are used as diluents for fluorine [84,87,88,91,92], but this inevitably results in carry-over of target product with by-products, that's why solvents with higher boiling point are used for dilution of initial substrate: anhydrous hydrogen fluoride [84,87,88], ice acetic acid [85], trichlorotrifluoroethane (freon 113) [20], water [90] and others.
The role of solvent at direct fluorination can't be unambiguously defined as ordinary dilution.
Thus, in the work [84] the theory of one-electron transfer is offered as starting stage of fluorination process, when compounds reacting between each other make up donor-acceptor systems. Then the process of substitutive fluorination at saturated carbon atom can be presented as scheme (23):
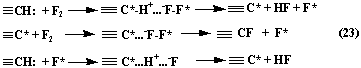
Following that the formation of radical particles occurs, but for providing process transferring out of radical into ionic the use of solvation effect is necessary, due to which radicals have time to re-combine until the leaving into reaction volume and chain reaction of fluorination is not realized. This allows to control direct fluorination process of different organic compounds, greatly simplifying the technological scheme.
Anions of acids, polynitrocompounds, amines and amides belong to compounds able to solvatate forming radicals.
By the example of tri-nitrile anion the scheme is possible (24):
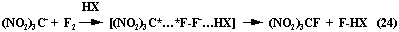
The high affinity of fluorine with electron is already realized at the initial stage in the form of electron transfer from partner's molecule and doesn't depend on characteristics of the last mentioned one [87,88].
The mechanism of concrete reaction is determined by totality of surrounding conditions, including solvation.
Usually, perfluorinated carbons are used for dilution, their fluorine atoms form hydrogen bonds with replaceable hydrogen atoms. Thus, in the work [92] perfluorohexane was used, and introduction of 100% fluorine was carried out at such rate, which make its concentration in the solution 0,02M. Partly fluorinated amines and ethers, obtained out of corresponding perfluoroolefines, were used as raw material.
In the work [91] the successful using of methylsulphonylfluoride direct fluorination using 30% fluorine-nitrogen mixture is described.
The temperature of process conducting was determined by used solvent:
For hydrofluoride +10oC;
For formic acid +30oC;
For perfluoroisohexane +40oC.
At that noticeable yield (up to 64%) of tri-ortho-methylsulphonylfluoride was obtained at using of perfluoroisohexane. It is important, that mono- and difluoromethylsulphonylfluorides were identified as admixtures, they were also fluorinated with the yield 88,8 and 97,1% respectively, that confirms the conclusions, made before in the works [82,83,84].
On the base of analysis of literature data we can see, that now there is no efficient production technology of perfluoroalkylsulphonylfluorides, which would be provided with available raw materials and construction materials.
The aim of the present work is searching and working out of PFSG efficient obtaining method using inexpensive domestically produced raw materials.
After considering obtaining methods of PFSG, we decided to research the opportunity of Ride's reaction (sulphochlorination) application to monohydroperfluoroparaffins:
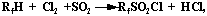
And also fluorination of monohydroperfluoroalkylsulpho fluorides, obtained by hydrolysis of corresponding sultones
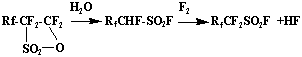
Sulphochlorination. In the work trifluoromethane and pentafluoroethane sulphochlorination was studied. At UV-irradiation (wave length 1950-3100 nm) of the mixture: paraffin, chlorine and sulfurous anhydride in propprtion 1:1:1 the formation of perfluoroalkylsulphochlorides is observed, though the rate of interaction is low and yield of target product doesn't exceed 10%. As we suppose, it is connected with high oxidization grade of paraffin molecule and screening effect of fluorine atoms. CF3SO2Cl was identified by IR-spectra [95].
Fluorination of monohydroperfluoroalkylsulphofluorides.
Fluorination of monohydroperfluoroalkylsulphofluorides was carried out both using elemental fluorine and cobalt trifluoride. At fluorination of C2F4HSO2Cl using cobalt trifluoride C2F4HSO2F is formed, however its yield doesn't exceed 20%. Other products of reaction are SO2F2, CF4, C2F6 and HF.
Fluorination of monohydroperfluoroalkylsulphofluorides using elemental fluorine appeared to be more successful. At interaction of CF3CHFSO2F with fluorine at 20-50oC C2F5SO2F was obtained with the yield close to 93%. Main admixtures are SO2F2, CF4, C2F6. This obtaining method of perfluoroalkylsulphofluorides was patented.
Refereces
1. Chemie und Technologie aliphatischer fluororganischer verbindungen, 1964, Studdgart, D. Osterroth
2. U. Sheppard. K. Sharts. Organicheskaya khimiya ftora (pod red. I.L. Knunyantsa) M.; «Mir», 1972
3. J. Saimons. «Ftor i ego proizvodnye», t. 1, Izdatinlit, 1953
4. Veigand-Higeltag. Metody ehksperimentov v organicheskoj khimii. M: «Khimiya», 1968, c. 574
5. M. Steisi, D.K. Tatlow. Uspekhi khimii ftora, t.1, M: «Khimiya», 1964
6. A. I. Rahimov. «Khimiya i tekhnologiya ftororganicheskih soedinenij», M: «Khimiya», 1986
7. Tomilov A.N. «Elektrohimicheskij sintez ftororganicheskih soedinenij», M: Goskhimizdat, 1956, s. 576-579
8. Isikawa N., Kabayasi E. «Ftor. Khimiya i primenenie», M: «Mir», 1982, c. 91
9. Isikawa N. «Soedineniya ftora. Sintez i primenenie», M: «Mir», 1990, s. 9-39
10. Patent 2915544 USA (1959)
11. Ryse I.G. «Khimiya ftora i ego neorganicheskih soedinenij» M: Goskhimizdat, 1956, s. 576-579
12. Patent 818756GB (1959).
13.Patent 0364340 EP (1990).
14. Nikanorov Yu.I., Karelin A.I. i dr. Primenenie interkalirovannyh v grafit kislot L'yuisa v kachestve katalizatorov reaktsij Fridelya-Kraftsa. IV Vsesoyuznaya konferentsiya po ftororganicheskim soedineniyam, Tashkent, 1982, s.12
15. Gida V.M.‚ Pouderovskij Yu.A.‚ Belfman A.L.‚ Sterlin S.R.‚ German L.S.‚ Knunyants I.L. Fiziko-himicheskie osnovy ispol'zovaniya PAV na osnove ftorangidridov perftor- ftorsul'foniloksaalkilkarbonovyh kislot.IV Vsesoyuznaya konferentsiya po FOS, Tashkent, 1982
16. Averbah K.O.‚ Goldin G.S.‚ Nekrasova L.A. Soli ftororganicheskih ehfirov sul'fokarbonovyh kislot, IV Vsesoyuznaya konferentsiya po FOS, Tashkent, 1982, s.23
17. Kondratenko N.V.‚ Kolomejtsev A.A, Ignat’ev N.V., Yagupolskij L.M. Soli geksakis(triftormetilsul'fonil)difenilmetana-1‚3-difenilpropena i tris(triftormetilsul'fonil)1rifenilmetana -novyjtipanionoidnyhkrasitelej.IV Vsesoyuznaya konferentsiya po FOS, Tashkent, 1982, s. 72
18. Patent DE 10126929A/BRD (2002)
19. Eleev A.F.‚ Fetisov V.I.‚ Sokol'skij G.A., Ob atsidifitsiruyushchej sposobnosti ftorsul'fonil'noj gruppy vo ftorsoderzhashchih SN-kislotah. IV Vsesoyuznaya konferentsiya po FOS, Tashkent, 1982, s. 172
20. Furin G.G., Fajnzil'berg A.A. Sovremennye metody ftorirovaniya organicheskih soedinenij. M. «Nauka», 2000, s.240
21. Patent 26878 UA (1999) [Litij tris-(pentaftorehtil)-triftorfosfat] v kachestve ehlektrolita dlya himicheskih istochnikov toka. Yagupol'skij L.M., Yagupol'skij Yu.L.‚ Pavlenko N.V. i dr.
22. Patent 27067 UA (2000) Tris(triftorehtoksisul'fonil)metanid litiya i tris(triftorehtoksisul'fonil)metan. Yagupol'skij Yu.L.‚ Saviia T.I. i dr.
23. Patent 2950317 USA (1960)
24. Zili Chen, Wennan Xiong, Preparation and application in asymmetric alkynylation of degehyds. China. J. Royal Society of Chemistry, 2002, p.2098-2099
25. Patent 2877267 USA (1959).
26. Patent 3542864 USA (1970).
27. Patent 2276097 USA (1942).
28. Temple S. The reaction of sulfuryl fluoride and sulfonyl fluoride with fluoroolefins. J. Organic Chem. 1968. v.33,N 1, p.344-346
29. Patent 2403207 USA (1946).
30. Koshar R.J. Trott P.W. La Zerte J.D. Preparation of b-H-perfluoroalkanesulfonic acids. J. Am.Chem.Soc., 1953(75), p.4595
31. Langlois B.R. Difluoromethanesulfonic acid. I An Improved route to sodium difluoromethanesulfonate.1990. J.Fluorine Chem. 46, p.407-421.
32. Furin G.G. Ftoristyj vodorod. Ftoriruyushchij agent i rastvoritel' v organicheskom sinteze.Novosibirsk, 2002, s. 335-342
33. Furin G.G. Novye aspekty primeneniya perftoralkilgalogenidov v sinteze ftoroorganicheskih soedinenij.Zh. UspekhiKhimii, 69 (6) 2000, s. 538-570
34. Benefice-Malounet S. Blancou H. et all. Mise au point D’un procede de preparation catalitique de perfluoralkyl Sulfonamide nar addition diethylamine a un chlorure d’acide perfluoralcane sulfonique. J.Fluorine Chem. 31,1986, p.319-332
35. Wei-Yuang, Jian-Long Chen at all. Reactions of perfluoroalkanesulfonyl bromide. J.Fluor. Chem. v.35, N1, 1987,19-21
36. Jin Tao Liu, GuO-Dong Sui, Gang Chem, Wei-Yuan Huang, Sodium ditionite initiated additions sulfination reaction of perfluoroalkyl bromides and olefins.J. Fluorine Chem. 93, 1999, 49-51
37. Fokin A.V., Rapkin A.I., Matvienko V.I. Izvestiya AN SSSR, ser. khim., 1985 (8), 1929
38. Patent 586666 Sw. (1977).
39. Knunyants I. L., Yakobson G.G. «Sintezy ftoroorganicheskih soedinenij.» -M. Khimiya, 1973, s. 76.
40. Knunaynts J.L. and Sokolski G.A. Fluorinatedβ-Sultones. Angew. Chem. Internat. Edit/ vol.11 (1972) N7, p.583-585
41. Dmitriev M.A., Sokol'skij G.A., Knunyants I.L., Ftorsoderzhashchie β-sultony. Izvestiya AN SSSR, ser.khim., 1960 (6), s. 1035-1038
42. Cheburkov Y, Lamanna W.M. Fluorinated olefins and oleum. J. Fluorine Chem. 2003, 121,147-152
43. Patent 4206138 USA (1980)
44. England D.C., Dictrich M.A., Lindsey. Reactions of fluoroolefins with SO3, J. Amer.Chem. Soc., 82, 1960, 6181
45.BeloventsevM.A.,MiheevL.L.,PavlovV.M.,SokolskijG.A.,KnunyantsI.L.,Reaktsiya (CF3)2C=CF2sSO3.Izvestiya AN SSSR, ser. khim., 1972 (11), s. 2510-2516
46.Patent 4567003USA (1986)
47. Patent 3689545 USA (1972)
48. Krylov I.I., Kutenov A.P., Sokolskij G.A. β-Sultony na osnove α,ω-perftordienov. IV Vsesoyuznaya konferentsiya po FOS, Tashkent, 1982, s. 180
49. Volkov M.D., Nazaretyan V.P., Fotoliz ftorsul'fonil diftoratsetilhlorida. Zh. Org. Khimii, t. XIII, v. 8 (1977), s. 1788-1789
50. Banks R.E., Binchall J.M., Haszeldine R.N. et all. Perfluoroallyl Fluorosulphonate. J. of Fluorine Chemistry, 20 (1982),1133-1134
51. Radchenko O.A., Ilchenko A.YA., Yagupolskij L.M. Sintez i reaktsii pentaftor ehtansulfinovoj i pentaftorsul'fokislot.Zh. Org. Khimii, t. XVII, v. 3 (1981)
52. Patent DE 19728560 A1, BRD (1999)
53. Patent 20341172, BRD (1972)
54. Patent 1443517, BRD (1968)
55. Patent 1096273 Fr. (1955)
56. Patent 4425199 USA (1984)
57. Patent 2234837, BRD (1974)
58. Matalin V.A., Shkultetskaya L.V., Moldavskij D.D., Kaurova P.N., Barabanov V.G. Issledovanie protsessa sinteza pentaftorehtansul'fonilftorida i ego proizvodnyh. // Tezisy dokladov 3-j mezhdunarodnoj konferentsii «Khimiya, tekhnologiya i primenenie ftorsoderzhashchih soedinenij v promyshlennosti», Sankt-Peterburg. June 3-6 2001, s.1-22
59. Patent 2732398 USA (1956)
60. Fauler R.D.‚ Berford H.B., Gamilton G.M. i dr. Sintez ftoruglerodov. «Khimiya ftora» s.6. №1 - M. IL, 1950, s. 91-113
61. Benner R.G., Benning A.F., Dounin F.B. i dr. Ftoruglerody, poluchennye ftorirovaniem trekhftoristym kobaltom. «Khimiya ftora», s.6. №1 —M. IL, 1950, s.114-128
62. Patent appl. 60-81134 Japan, Poluchenie oktaftorpropana. Katamura Konti, Kageyama Yutaka, Nakalma Hidetosi // Publ. 09.05.1985
63. Steisi M.‚ Tatlow D.K. «Uspekhi khimii ftora» T1, - M: «Khimiya», 1964, s.424-471
64. Kornilov V.V., Kostyaev P.A., Maksimov B.N., Melnichenko B.A., Fedorova T.E. Ftorirovanie organicheskih soedinenij triftoridom kobalta / Zh. Prikladnoj Khimii. t.68‚ v.9‚ 1995, s.1409-1418
65. Asovich V.S.‚ Kostyaev P.A. Makrokineticheskie zakonomernosti protsessa ftorirovaniya geksaftorpropena triftoridom kobalta/ Zh. Prikladnoj Khimii, t.67, v.8‚ 1994, s. 1320-1323 ; 65-1. Kostyaev P.A., Pashkevich D.S. Rol' ftorovodoroda i diftorida kobal'ta v protsessah ftorirovaniya uglevodorodov triftoridom kobalta/Zh. Prikladnoj Khimii, t.67, v.12, 1994, s.2012-2016
66. Bispen T.A.‚ Borutskaya G.V.‚ Mihailova T.V.‚ Moldavskij D.D.‚ Furin G.G. Poluchenie i svojstva nekotoryh poliftorirovannyh pentanov /Zh. Prikladnoj khimii, t.68, v.5, 1995, c.793-796
67. Bispen T.A., Mihailova T.V., Moldavskij D.D.‚ Furin G.G.‚ Shkultetskaya L.V. K voprosu ob ochistke perftororganicheskih soedinenij / Zh. Prikladnoj Khimii, t.69‚ v.1, 1996, s.112-119
68. Moldavskij D.D.‚ Shkultetskaya L.V., Furin G.G. Sravnitel'noe ftorirovanie organicheskih soedinenij CoF3 kak ftoriruyushchim agentom i CoF3 v prisutstvii ehlementnogo ftora/ 3-ya mezhdunarodnaya konferentsiya po khimii perftororganicheskih soedinenij. Sankt-Peterburg, 6-9.06.2001, Obzor.
69. Asovich B.C., Prokudin I.P. Osobennosti ftorirovaniya organicheskih veshchestv chetyrekhftoristym tseriem.
70. Patent 4143079 USA (1979)
71. Patent 2831035 USA (1958)
72. Patent 3709800 USA (1973)
73. Keid D.I., Grosse A.V., Barbor E. Poluchenie ftoruglerodov kataliticheskim ftorirovaniem uglevodorodov / «Khimiya ftora», sb. № 1, M, IL, 1950, s. 129-135
74. Bigelou L.A. Dejstvie ehlementarnogo ftora na organicheskie soedineniya // «Ftor i ego soedineniya», pod. red. J. Saimons, t. 1, M, IL, 1953, s. 314-335
75. Patent 4113453 USA (1978)
76. Patent 4281119 USA (1981)
77. Patent 4330475 USA (1982)
78. Zaharov V.Yu., Denisov A.K., Novikova M.D. Pryamoe gazofaznoe kataliticheskoe ftorirovanie ne polnost'yu ftorirovannyh organicheskih soedinenij. / Zhurnal organicheskojkhimii, t. 30, v. 12 s. 1844-1846
79. Ruff J.K. The catalytic fluorination of perfluorocarbon nitriles and imines/ J. Org. Chem., 1967, v.32,N5, p.1675-1677
80. Honorat F.A., Shreev J.M. Bis-(fluoroxy)-difluoromethane CF2(OF2). J. Amer. Chem. Soc.,1967,v89, N8, p.1809-1819
81. Lustig M.A., Pitochelli A.R., Ruff J.K. The catalytic addition of fluorine to a carbon group. Preparation of fluoroxy compounds // J. Amer. Chem.Soc. 1967,v.89,N12,p.2841-2843
82.BispenT.A.‚MoldavskijD.D.‚FurinG.G.Kproblemetekhnologiipolucheniyaperftororganicheskihsoedinenij/Zh. PrikladnojKhimii. 1998, t 71, v.8, s.1334-1337
83. Bispen T.A., Moldavskij D.D., Furin G.G. Poluchenie perftorirovannyh parafinov, prostyh ehfirov i tretichnyh aminov pryamym ftorirovaniem ehlementarnym ftorom/ Zh. PrikladnojKhimii, 1998, t.71, v.6, s.977-981
84. Furin G.G. Gambaretto G.P. Direct fluorination of organic compounds. Padova:CLEUP 1996, p.83-86, 112
85. Jtsumaro Kumadaki, Masakazu Nakazawa and Yochiro Kobayashi. Direct fluorination of 6-O-Cycloyracil Nucleosides// Tetrahedron Letters, vol.24, N10, p.1055-1056 (1983)
86. Patent 3718627 USA (1973)
87. Furin. G.G. Ftoristyj vodorod. Ftoriruyushchij reagent i rastvoritel' v organicheskom sinteze. Pryamoe ftorirovanie. Novosibirsk, 2002, s. 138-170
88. Muhametshin F.H. Perenos ehlektrona kak nachal'naya stadiya reaktsij ehlementarnogo ftora // Zh. Prikladnoj Khimii, 1997, t.70, v.1, s.127-134
89. Chiracal R.Firrau, Garnett E.S. Direct elemental fluorination of tyrosines in HF/BF3. Reactivity and Selectivity. J.Fluorine Chem., 1994, vol.45, p.125
90. Diksic M and Di Raddo P. New synthesis of fluoro-compounds by fluorination in water./ Tetrahedron Letters, vol.25, N 43, p.4885-4885, 1984
91. Masafumi Cobayashi, Tetsuya Jnoguchi, Takashu Jida et all. Development of direct fluorination technology for application to materials for lithium battery. J. Fluorine Chem. 2003, 120, p.105-110
92. Scherer K.V., Kouchi Yamanauchi and Taizo Ono. A new syntetic approach to perfluorochemicals: photofluorination with 100% fluorine in solution. J. Fluorine Chem 1982, vol.21, N1, p.48
93. Sasaki Yukio, Ebara Ryo, Nanbi Noritoshi, Takenaro Masahiro. Direct fluorination ?-butirolaktona/ J. Fluorine Chem. 2001, 108, N11, p.117-120
94. Banks R. Eric, Murtagh Vincent et all. Direct fluorination bis-(trifluoromethanesulfonil)-imide and derivatives. J. Fluorine Chem. 2001, 112, N2, p. 271-275
95. Hu L.Q., Des Marteau D.D. Inorg.Chem., 1993,v.32, p.5007-5010
Fluorine Notes, 2004, 36, 3-4
