Fluorine Notes, 2002, 24, 3-4
Fluorine notes, Vol. 5(24) 2002
|
|
Rf |
X | R | T melt oC |
IR spectra cm-1 |
| CF3 "À-1" | H | H | 103-105 | 3400; 3220; 3080-2900; 1710; 1630; 1610; 1605; 1580; 1335; 1315; 1230; 1205; 1170; 1155; 1060; 1030; 1000; 975; 965; 930; 850; 760; 740; 695; 660; 640; 530 |
| CF3 "À-2" | p-Cl- | H | 227 (decomp.) | 1680; 1630; 1600; 1595; 1580; 1480; 1320; 1210; 1180; 1095; 1020; 975; 940; 870; 835; 830; 730; 560; 505 |
| CF3 "À-3" | p-NO2 | H | 254 (decomp.) | 1685; 1635; 1600; 1530; 1295; 1215; 1110; 1025; 985; 980; 960; 860; 840; 750; 695; 560 495 |
| CF3 "À-4" | p-HO- | H | 198 (decomp.) |
3490; 1680;5; 1675; 1625; 1610; 1600; 1565; 1515; 1505; 1300; 1285; 1260; 1235; 1205; 1165; 1130; 1025; 970; 960; 945; 890; 840; 820; 785; 720; 555; 510; 490 |
| CF3 "À-5" | p-(CH3)2-N- | H | 79-82 | 1730; 1675; 1665; 1605; 1590; 1550; 1530; 1360; 1320; 1250; 1235; 1205; 1190; 1180; 1130; 1070; 950; 825; 730; 525 |
| C4F9"À-6" | H | CF3 | 78-80 | 3434; 3040-2920; 1970; 1670; 1605; 1566; 1493; 1444; 1363; 1311; 1286; 1178; 1163; 1075; 1023; 915; 760; 690; 652; 565 |
TABLE 2
CHARACTERISTICS OF PRODUCTS OF DEHYDRATION OF PERFLUOROACYLHYDRAZONES
|
Initial substance |
Product | Ò melt., 0C | IR spectra, cm-1 | Spectra 1Í, md DMCO-d6 |
Spectra 13C, md |
| "À-1" | "B-1" | 67-69 | 3200, 1710, 1670, 1625, 1575, 1305, 1210, 1170, 1150, 1070, 1020 | 7,50; 7,51; 7,88; 7,89; 8,72 | 127,62; 128,29; 128,83; 131,29; 161,43 |
| "À-2" | "A-2" | - | 1680; 1630; 1600; 1595; 1580; 1480; 1320; 1210; 1180; 1095; 1020; 975; 940; 870; 835; 830; 730; 560; 505 | - |
- |
| "À-3" | "A-3" | - | 1685; 1635; 1600; 1530; 1295; 1215; 1110; 1025; 985; 980; 960; 860; 840; 750; 695; 560 495 | - |
- |
| "À-5" | "B-5" | 208-211 | 3200; 1700; 1605; 1550; 1520; 1300; 1230; 1185; 1170; 1065; 960; 950; 820; 750; 730; 530 | 6,7; 6,72; 7,25; 7,68; 7,70; 8,57 | 111,72; 122,30; 129,81; 152; 10; 160;67 |
EXPERIMENT
NMR spectra 1H and 13C of the substance solutions in DMSO-d6 were recorded with the help of Bruker AM 500 NMR spectrometer (work frequencies
were 500MHz and 75MHz respectively), the chemical shifts are shown relative to
TMS. IR spectra of the samples suspension in vaseline oil were recorded with
the help of IR-spectrophotometer Specord IR 75.
Hydrazide of trifluoroacetic
acid was prepared from ethyl ester of trifluoroacetic acid [2] in line with the
published methods [3, 4].
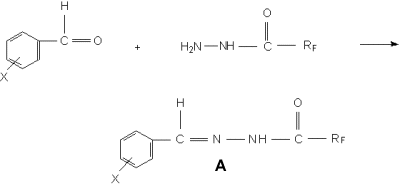
N,N-Trifluoroacetylbenzalhydrazone ("A-1"). The solution prepared of 2,2 g (0,02 mole)
of benzaldehyde in 5 ml of alcohol was added to 2,56 g (0,02 mole) of trifluoroacetylhydrazine
in 45 ml of alcohol. The blend was mixed for 2 hours at room temperature. The
blend was allowed to stand for a night till the sediment disappeared. The solvent
was removed until dry under vacuum of a water-jet pump. The obtained product
was 4,1 g (95%) benzalhydrazide (4 á), yellowish-brown powder. Rf 0,10 (toluene).
Cyclocondensation of N,N-Trifluoroacetylbenzalhydrazone. To the suspension of 2,16
g (0,01 mole) benzalhydrazide of trifluoroacetic acid in 25 ml of chloroform
we added the solution prepared of 1,1 ml of phosphorus oxychloride in 5 ml of
chloroform under continuous blending during 30 min. The resulted blend was heated
to 70 0C and allowed to stand for 5 hours. Then the solvent was distilled,
and the residue was dried under vacuum, treated with 70 ml of water, and neutralized
with sodium bicarbonate. The product was filtered, washed with water in the filter
(2*20 ml), and dried in the air. The obtained product was "B-1" 0,9 g (46 %),
yellow powder. Rf 0,50 (toluene).
Calculated: C 54,55; H
1,86; F 28,79. C9H5F3N2.
Obtained: C 54,67; H 2,03; F 28,7.
IR
spectrum, vaseline oil (cm-1): 3200, 1710, 1670, 1625, 1575, 1305,
1210, 1170, 1150, 1070, 1020.
NMR 1Í, DMSO -d6 (md):
7,50; 7,51; 7,88; 7,89; 8,72.
NMR 13C, DMSO-d6 (md): 127,62; 128,29; 128,83; 131,29; 161,43.
Mass spectrum: m/z 208
[M+].
N,N-Trifluoroacetyl(ð-õëîðbenzal)hydrazone ("A-2"). The solution of 3,5 g (0,025
mole) of ð-chlorobenzaldehyde in 20 ml of alcohol was added to the solution of
3,2 g (0,025 mole) of trifluoroacetylhydrazine in 10 ml of water under continuous
mixing at room temperature. The blend was mixed for 2 hours and allowed to stay
for a night. The drained light-lemon sediment was dried in the air. The yield
was 3,6 g (57,4%). Rf 0,66 (toluene); 0,80 (toluene/chloroform = 4:1).
N,N-Trifluoroacetyl(ð-nitrobenzal)hydrazone ("A-3"). Under similar conditions
(see "A-2") from 4,35 g (0,03 mole) of ð-nitrobenzaldehyde and 3,48 g (0,03 mole)
of trifluoroacetylhydrazine we obtained 4,7 g (60%) of light-yellow crystal powder.
N,N-Trifluoroacetyl(ð-oxybenzal)hydrazone ("A-4"). Prepared at the same manner
as ("A-3") from 4,20 g (0,034 mole) of ð-oxybenzaldehyde and 4,9 g (0,039 mole)
of trifluoroacetylhydrazine. The yield was 4,6g (65,7%).
N,N-Trifluoroacetyl(ð-dimethylaminebenzal)hydrazone ("A-5"). The solution of 4,2 g
(0,03 mole) of trifluoroacetylhydrazine in 25 of alcohol was added to the solution
of 4,5 g (0,03 mole) of ð-dimethylaminebenzaldehyde in 50 ml of alcohol under
continuous mixing during 30 min. The blend was allowed to stay for 24 hours at
room temperature, after that 50 ml of water was added. The sediment was drained
and dried. The yield was 6,0 g (77 %), Ò.melt. 79-820C.
Cyclocondensation of N,N-trifluoroacetyl(ð- dimethylaminebenzal)hydrazone. The solution
prepared of 3 g (0,02 mole) of phosphorus oxychloride in 5 ml of carbon tetrachloride
was added to the suspension prepared of 1,56 g (0,006 mole) of N,N-trifluoroacetyl-(ð-dimethylaminebenzal)hydrazone
("A-5") in 25 ml of carbon tetrachloride under continuous mixing during 30 min.
The reaction blend was boiled for 14 hours under stirring. The solvent was distilled,
the residue was treated with 30 ml of alcohol and then with 20% solution of sodium
carbonate until ðÍ=8. The sediment was drained and dissolved in 30 ml of chloroform.
The solvent was evaporated. The product was re-crystallized from alcohol. The
yield was 0,7 g (47%); light brown powder, Ò.melt. was 208-2110C.
NMR 1Í, CDCl3 (md.): 7,24; 7,44; 7,45; 7,94; 7,95; 7,96.
NMR 13C, CDCl3 (md): 126,63; 128,35; 129,60; 138,50; 157,74.
Mass-spectrum: m/z 294 [M+].
N,N-Tridecafluoroheptanoyl-(ð-dimethylaminobenzal)hydrazone. The solution prepared
of 2,0 g (0,0053 mole) of hydrazide of perfluoroenanthoil acid (T melt. 850C)
in 25 ml of alcohol was added to the suspension prepared of 0,75 g (0,005 mole)
of ð-dimethylaminobenzaldehyde in 15 ml of alcohol under stirring at room temperature,
then heated to 45-500C and allowed to stay for 4 hours under continuous
stirring, and for a night at room temperature. Crystal sediment was formed. We
added 50 ml of water under stirring. The formed sediment was drained and dried.
The yield was 2,2 g (85%); Ò melt. 96-980C. C16H12F13N3O.
N,N-Nonafluoropentanoyl-(ð-dimethylaminobenzal)-hydrazone. Prepared at the
same manner from 0,005 mole of ð-dimethylaminobenzaldehyde and hydrazide of perfluoropentane
acid.
N,N- Nonafluoropentanoyl -(2,4-dimethoxybenzal)-hydrazone
Acetophenone
trifluoroacetylhydrazone ("A-6"). Prepared at the same manner as ("A-4") from
3,6 g (0,03 mole) of acetophenone and 4,2 g (0,03 mole) of trifluoroacetylhydrazine
with the yield 3,1 g (44 %).
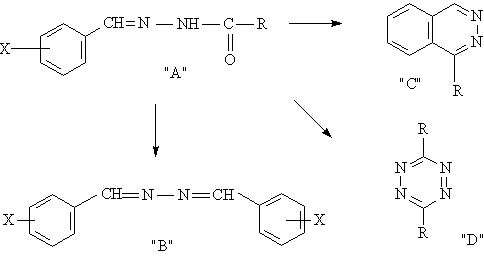
CONCLUSIONS
1. A number of perfluoroacylhydrazones of aromatic aldehydes is synthesized.
2. It is determined that perfluoroacylhydrazones with electron-seeking substituents
in aromatic fragment are chemically stable under the experiment conditions.
3. It is shown that dehydration of perfluoroacylhydrazones with electron-donating
substituents occurs with splitting-off of perfluoroacyl fragment and results
in formation of benzylidene-azine derivatives.
References
2. Gudlitskij M. Khimiya organicheskih soedinenij ftora. M.: GNTIHL, 1961, s. 174.
3. H.C. Brown, M.T. Cheng, L.T. Parcell, D.Pilipovich / J. Org. Chem. 1961. V. 26, N 11. P. 4407-4410.
4. Mazalova Z.I.‚ Lopyrev B.A. / Zh Org. Khimii. 1971. T. 7, V. 7. s. 1408-1410.
Fluorine Notes, 2002, 24, 3-4
