Fluorine Notes, 2001, 14, 3-4
Trimethyl(pentafluorophenyl)silane |
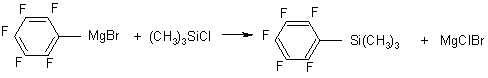
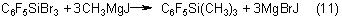
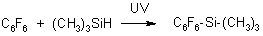
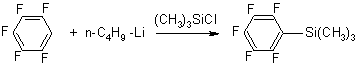
| N | Initial carbonyl compound | Products produced | Yield, medium | References |
| 1 | 2 | 3 | 4 | 5 |
| 1 | Diacetoamide(equimolar quantity) | N[1(trimethylsiloxy)ethylidene]acetamide | Diethyl ether | 26,27,28 |
| 2 | N-trimethylsilylsuccinamide | 2,5 bis(trimethylsiloxy)-N-trimethylsilylpyrrole | 28 | |
| 3 | mathylmesitylketone | 1,1-mesityltrimethylsiloxyethene | THF | 29 |
| 4 | 3-pyridine aldehyde | Mixture of pyridil(pentafluorophenyl)trimethylsiloxy
methane and è 1,2-bis(trimethylsiloxy)-1,2-bis (3-pyridil)ethene |
Diethyl ether
45% 16% |
29 |
| 5 | Trifluoro-substituted acetophenone | 1-phenyl-1-pentafluorophenyl-1-trimethylsiloxy- 2,2,2-trifluoroethane | Acetonitrile
85% |
30,33 |
| 6 | Acetophenone | 1-phenyl-1-trimethylsiloxyethene | Diethyl ether
81,2 % 68,1 % |
23 24 |
| 7 | p-aminoacetophenone in the ratio of 1:1 to silane | 4-N-trimethylsilyl aminoacetophenone | MeCN
69,8 % |
33 |
| 8 | p-aminoacetophenone in the ratio of 1:2 to silane | 1-(4-trimethylsilyl-aminophenyl)-1-trimethylsiloxyethene | MeCN
85 % |
32,33 |
| 9 | 4 -nitroacetophenone | 1-(4-nitrophenyl)-1-trimethylsiloxyethane | MeCN
82-84 % |
33 |
| 10 | 2,3,4,5,6-pentafluoroacetophenone | 1-pentafluorophenyl-1-trimethylsiloxyethene | Diethyl ether
66 % |
33 |
| 11 | 2,4,6-trimethyl-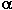 , , -trifluoroacetophenone , , -trifluoroacetophenone |
Tetrafluoropolyphenylene | THF
98 % |
33 |
| 12 | 2,2-dimethylbutanone | 3,3-dimethyl-2-trimethylsiloxybutene | Diethyl ether
88,6 % |
23,24 |
| 13 | Cyclohexanone | 1-trimethylsiloxy-cyclohexene-1 | Diethyl ether
36,5 % |
23,24 |
| 14 | Benzaldehyde | Phenyl( trimethyl-siloxy)pentafluorophenylmethane | Diethyl ether
56,5 % |
23 |
| 15 | 1-hexine-3-one | 3-trimethylsiloxy-3-hexene-1-ine | Diethyl ether | 24 |
| 16 | 2-methylpropanal | 1-pentafluorophenyl-1-trimethylsiloxy-2-methylpropane | 37,3 % | 24 |
The reaction of silane with carbonyl compounds is also catalyzed with fluoride ions, which sources are KF, CsF, RbF.
The synthesis of polyfluoroaromatic ethers is made through an intermediate polyfluoroalkooxide, a product of interaction of a perfluorocarbonyl compound with silane, which reacts in situ with pentafluorobenzyl bromide in 40% yield..
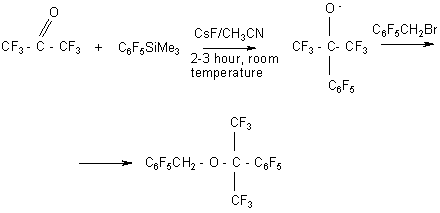
At the same time in the reaction of bis(trifluoroethyl) carbonate with silane in the presence of CsF there is formed the intermediate compound
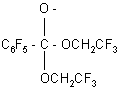
which generates CF3CH2O-anion. The latter substitute the fluorine atom in the pentafluorophenyl group in para position. In this case there is no interaction with pentafluorobenzyl bromide and diether is formed in 27% yield (35).
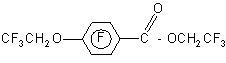
Paper (36) describes the addition of pentafluorophenyl group to benzaldehyde in the presence of TASF catalyst (tris(diethylamino)sulfonium difluorotrimethylsilane ((R2N)3S+Me3SiF2-) in a THF medium. In the author's opinion, the yield of phenyltrimethylsiloxy pentafluorophenylmethane with the use of this catalyst is higher (87%) than that with use of different catalysts.
2.2. Interaction of silane with a number of other non-carbonyl electrophilic compounds.
The interaction of compounds containing S,O and the active S-Cl(F) bond with silane was carried out in the presence of KF and TBAF(n-tetrabutylamino fluoride) catalysts. The following pentafluorophenylsulfoxides and pentafluorophenylsulfones were produced in a 75-95% yield:
- Pentafluorophenylsulfinyl fluorides from SOCl2 and SOF2
- Pentafluorosulfinyl fluorides from SO2Cl2 and SOF2
- Trifluoromethyl(pentafluorophenyl)sulfoxides and trifluoromethyl (pentafluorophenyl) sulfones from CF3S(O)F and CF3SO2Cl respectively (37).
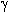 - SO3 freshly made also readily reacts with silane to form C6F5SO2OSi(CH3)3 in 69% yield (38).
- SO3 freshly made also readily reacts with silane to form C6F5SO2OSi(CH3)3 in 69% yield (38).
CsF was found an effective catalyst in the reactions with electrophilic compounds (39):
| Initial product | Compound obtained | Yield, % |
| I2 | Iodopentafluorobenzene | 59 |
| Br2 | Bromopentafluorobenzene | 93 |
| D2O | Deuteropentafluorobenzene | 38 |
| CH3I | Pentafluorotoluene | 59 |
| C6F5CF3 | perfluoro-4-methyldiphenyl | 50 |
| C6F5HgOCOCF3 | Perfluorodiphenyl mercury | 62 |
| IF5 | Pentafluorophenyliodide tetrafluoride (40) |
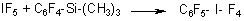
Reactions of silane with HF, AlCl3, AlBr3, HCl-AlCl3, Br2-AlBr3 in a medium of halohydrocarbons have been studied (41).
2.3. Interaction of silane with electrofilic compounds containing multiple bonds.
Silane reacts with polyfluoroalkenes in acetonitrile in the presence of CsF with substitution of the vinyl atoms of fluorine with the pentafluorophenyl group. The interaction of silane with perfluoro-4-methyl-2-pentene results in formation of perfluoro-4-methyl-2-phenyl-2-pentene in equal amount together with perfluoro-1,1,3-trimethylindane (42).
Fluorine-containing ethylene compounds in the presence of CsF react with silane with substitution of the fluorine atom at the 1 position with pentafluorophenyl group. As a result of the reaction, 7,8-dichloroperfluoro-1-octene converts into cis,trans-7,8-dichloroperfluoro-1-phenyl-1-octene in 84% yield. The side chain of perfluoroallylbenzene is arylated to form cis and trans perfluoro-1,3-diphenylpropene in 39% yield. In perfluoroisobutene the both vinyl atoms of fluorine are subsequently substituted to form perfluoroisobutenylbenzene, perfluorodiphenylisobutene and perfluoro-4-isobutenyl-diphenyl, a product of fluorine substitution in the pentafluorophenyl ring of perfluoroisobutenylbenzene:
CF2=C(CF3)2 + C6F5Si(CH3)3  C6F5-CF=C(CF3)2
(33 %) +
C6F5-CF=C(CF3)2
(33 %) +
+ (C6F5)2C=C(CF3)2 (7 %) + 4- C6F5C6F4 CF=C (CF3)2 (10 %)
Internal perfluoroalkenes react with silane under more hard conditions: at 55-60oC. So, perfluoro-2-methyl-2-pentene is arylated into 2-methyl-3-phenyl-2-pentene in 63% yield (43). Studying the reactions of silane with internal perfluoroalkenes, the authors of (44) have found that azoalkenes, perfluoro-3-azapentene-2 and perfluoro-azanonene-4 in the presence of CsF at 20oC are converted into appropriate perfluorophenylazaalkenes in 63% and 56% yield respectively.
Azenes in a medium of acetonitrile in the presence of CsF at 25oC form pentafluorophenyl substituted azenes under the influence of silane.
| Initial azene | Product produced | Yield, % |
| CF3(C2F5)NN=C(Cl)CF3 | CF3(C2F5)NN=C(CF3)-C6F5 | 65 |
| (CF3)2NN=C(Cl)C2F5 | (CF3)2NN=C(CF3)C6F5 | 68 |
| SF5N=C(Cl)C2F5 | SF5N=C(C2F5)C6F5 | 55 |
Sulfimides react with silane readily as well.
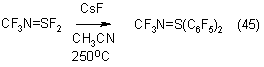
2.4. Interaction of silane with nucleophilic compounds.
In the study of the reactions of silane with N-,O-,S-,Si- and C- nucleophiles on examples of sodilum ethylate, sodium propanethiolate, piperidine, piperidyllithium, butyllithium, lithiumalumohydride and triphenylsilyllithium (Ph3SiLi) the authors have concluded that the nature of a nucleophile is the determining factor in the course of these reactions. Thus, O- and S- nucleophiles attack the Si atom whereas Si- and C-nucleophiles, lithium alumohydride and piperidyllithium attack C-4 atom of the pentafluorophenyl group and piperidine affects both centers (46,47).
Similar results in the reactions of n-butyllithium (48) and triphenylsilyllithium (49) were obtained earlier: butyllithium in hexane reacted with silane to give 1-(trimethylsilyl)n-butyltetrafluorobenzene in 54% and triphenylsilyllithium in THF gave 1-(trimethylsilyl)-4-(triphenylsilyl)-tetrafluorobenzene in 42.5% yield.
2.5. Interaction of silane with xenone difluoride
A reaction of silane with XeF2 in acetonitrile in the presence of fluoride ions (CsF, KF, RbF) results in evolution of xenone, trimethylfluorosilane and formation of pentafluorobenzene (58%) and decafluorodiphenyl (11%) (50,51). Silane does not react with XeF2 in fluorotrichloromethane, sulfuryl fluoride even in the presence of CsF.
The reactivity of xenone difluoride increases sharply in methylene chloride in the presence of boron trifluoride due to polarization of the Xe-F bond and already at room temperature silane is converted into 1-trimethylsilylheptafluoro-1,4-cyclohexadiene in 74% yield (52).
2.6. Other silane reactions.
The reaction of phenylacetylene with silane in the presence of a cyanide-ion is described in paper (25):
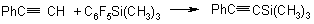
Silane reacts with potassium hexafluorobromate according to the following equation:
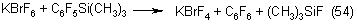
Silane was used for the production of anionic complexes of tetrakis(pentafluorophenyl)indium [In(C6F5)4]-by interaction of In(C6F5)3 * D, where D= MeCN, ET2O, with silane in the presence of CsF in acetonitrile (55,56).
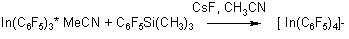
References
1. M. Fild, O. Glemser, G. Chritopf, Angew. Chem., 1964, v. 76, N 23, s. 953; Angew. Chem. Internat. Edit, 1964, v. 3, N 12, s. 801
2. J. M. Birchall, W. M. Daniewski, R. N. Haszeldine, L.S. Holder, J. Chem. Soc., 1965, p. 6702-6707
3. A.E. Jukes, H. Gilman, J. Organometal. Chem., 1969, 17 (1), p.145-148
4. J. Oliver, W. A. G. Graham, J. Organoelemental Chem., 1969, 19(1), p. 17-27
5. Н.С.Вязанкин, О.А. Вязанкина, Б.А. Гостевский и др. Ж. Общ. Химии, 1984, т. 54, вып. 2, с. 461
6. S.P. Kotun, I. D. O. Anderson, D.D. Des Marteau, J. Org. Chem, 1992, v. 57, p. 1124
7. G.G. Furin, O. A. Vyazankina, B.A. Gostevsky, N.S. Vyazankin, Tetrahedron, 1988, v.44, N 10, p. 2675
8. G. K. S. Prakash, A. K. Yudin, Chem. Rev., 1997, v. 97, n 3, p. 782-783
9. M. Schmeier, N. Wessel, M. Weidenbruch, Chem. Ber., 1968, v. 101, helt. 6, s. 1897
10. G. M. Brooke, R. D. Chambers, I Heyes, W. K. R. Musdrave, J. Chem. Soc., 1964, p. 729
11. M. Weidenbruch, N. Wessal, Chem. Ber., 1972, v. 105, N 1, s. 173-187
12. R. Fields, R. N. Haszeldine, A. E. Hubbard, J. Chem. Soc., C., 1979, N 16, p. 2193
13. D. Hebich, F. Effenberger, Synthesis, 1979, N 11, p. 841
14. F. Janzen and others, Chem Commun., 1966, 19, p. 672-673
15. V. V. Bardin, L. S. Pressman, L. N. Rogoza, G.G. Furin, Zh. Obshch. Khim., 1999, t. 62, T 10, 2342-2349
16. V. V. Bardin, L. N. Oparina, G.G. Furin, Izv. AN SSSR, ser. khimich., 1989, T 9, s. 2153-2154
17. Kuppert, K. Schlich, W. Volbach, Tetrahedron Lett., 1984, v. 25, N 21, p. 2195-2198
18. V. V. Bardin, L. S. Pressman, L. N. Rogoza, G.G. Furin, J. Fluor. Chem., 1991,v. 53,N 2, 213-231
19. V.V. Bardin, L. S. Pressman , G.G. Furin, Sibirskij khimicheskij zhurnal, 1992, N 3, s. 52-55
20. J. Grobe, Synlett., 1995, N 6, p. 641
21. A.F. Webb, D.S. Sethi, H. Gilman, J. Organomet. Chem., 1970, v. 21, N 2, p. 61-62
22. O.A. Reutov, Tetrahedron, 1978, v. 34, N 19, p. 2827
23. B.A. Gostevskij, O.A. Kruglaya, N.S. Vyazankin, Izvestiya AN SSSR, ser. khim., 1978, N 10, s. 2425
24. O.A. Kruglaya, V.A. Gostevskij, I. D. Kalihman, N.S. Vyazankin, Zhurn.Obshch.Khimii, 1979, t. 49, N 2, s. 354-360
25. A. Gostevskii, O.A. Kruglaya, A. I. Albanov, N.S. Vyazankin, J. Organomet. Chem., 1980,v. 187,p. 157-166
26. B.A. Gostevskij, O. A. Vyazankina, I. D. Kalihman, O. B. Bannikova, N.S. Vyazankin, Zhurn. Obshch.Khimii, 1983, t. 53, N 1, s. 229
27. I. D. Kalihman, B.A. Gostevskij, O. B. Bannikova, N.S. Vyazankin, O. A. Vyazankina, Izvestiya AN SSSR, ser.khim, 1983, N 7 s. 1515-1518
28. B.A. Gostevskij, O. A. Vyazankina, I. D. Kalihman, O. B. Bannikova, N.S. Vyazankin, Zhurn. Obshch.Khimii, 1983, t. 53, N 9, s. 2051-2054
29. B.A. Gostevskij, O. A. Vyazankina, N.S. Vyazankin, Zhurn. Obshch. Khimii, 1984, t. 54, N 5, s. 1209-1210
30. B.A. Gostevskij, O. A. Vyazankina, N.S. Vyazankin, Zhurn. Obshch. Khimii, 1984, t. 54, N 11, s. 2613-2617
31. B.A. Gostevskij, I. D. Kalihman, O. A. Vyazankina, N.S. Vyazankin, O. B. Bannikova, Zhurn.Obshch.Khimii, 1984, t. 54, N 6, s. 1428-1429
32. O. A. Vyazankina, B.A. Gostevsky, N.S. Vyazankin, J. Organomet. Chem., 1985,v. 292,N 1-2,p. 145-149
33. O. A. Vyazankina, N.S. Vyazankin, B.A. Gostevskij, Izv.AN SSSR ser. khim., 1985, t. 11, s. 2585-2589
34. O. A. Vyazankina, N.S. Vyazankin, B.A. Gostevskij, I. A. Titova, V. A. Lopyrev, I. D. Kalihman, Zhurn.Obshch. Khimii, 1984, t. 54, N 2, s. 461
35. M. Nishida, A. Vij, R. Kirchmeier, I. M. Shreeve, Inorg. Chem., 1995, v. 34, N 24, p. 6085-6092
36. M. Fujita, M. Obayashi, T. Hiyama, Tetrahedron, 1988, v. 44, N 13, p. 4135-4145
37. N. R. Patel, R. L. Kirchmeier, Inorg. Chem., 1992, v.31, p. 2537-2540
38. H. Holfter, R. L. Kirchmeier, I. M. Shreeve, Inorg. Chem., 1994,v.33,N 26,p. 6369- 6372
39. V.V. Bardin, I. V. Stennikova, G.G. Furin, Zhurn. Obshch. Khimii, 1988, t. 58, N 4, s. 812
40. H. I. Frohn, Chem.-Ztg., 1984, v.108, N 4, s. 146-147, CA, 1984, v. 101, 110434
41. H. I. Frohn, A. Lewin, V.V. Bardin, J. Organomet. Chem., 1998,v. 570,N 2,p. 255-263
42. V.V. Bardin, I. V. Stennikova, V. A. Petrov, G.G. Furin, L.S. German, Izv. AN SSSR, ser. Himicheskaya, 1989, N 2, s. 477-479
43. V.V. Bardin, V. A. Petrov, I. V. Stennikova, G.G. Furin, Zhurn. Org. Khimii, 1989, t. 25, N 1, s. 52-55
44. V. A. Petrov, V.V. Bardin, V.K. Grinevskaya, E.I. Mysov, G.G. Furin, L.S. German, Izv. AN SSSR, ser. khim., 1990, N 4, s. 923N.
45. R. Patel, R. L. Kirchmeier, I. M. Shreeve, Inorg. Chem., 1993,v. 32,N 22,p. 4802-4806
46. V.V. Bardin, I. V. Stennikova, G.G. Furin, Zhurn. Obshch. Khimii, 1987, t. 57, N 7, 1580-1583
47. V.V. Bardin, L.N. Rogoza, I. V. Stennikova, G.G. Furin, J. Fluor.Chem., 1992, v.59, N 2, p. 165-177
48. F.W.G. Fearon, H. Gilman, J. Organomet. Chem., 1967, v. 10, N 3, p. 535-537
49. F.W.G. Fearon, H. Gilman, J. Organomet. Chem., 1968,v.13, N 1,p. 73
50. V.V. Bardin, I. V. Stennikova, G.G. Furin, G.G. Yakobson, Zhurn. Org. Khimii, 1985, t. 21, N 2, s. 458-459
51. V.V. Bardin, I. V. Stennikova, G.G. Furin i dr., Zhurn. Obshch. Khimii, 1988, t. 58, N 11, s. 2580-2588
52. V.V. Bardin, H. I. Frohn, J. Fluor. Chem., 1993, v. 60, N 2-3, p. 141-151
53. V.V. Bardin, H. I. Frohn, J. Fluor. Chem., 1998, v. 90, N 1, p.93-96, CA, 1998, v. 129, 175385
54. W. Breuer, H. I. Frohn, Z. Anorg. Allg., Chem., 1992, 611, s. 85-91
55. Choi Zel-Ho, J. Korean. Chem. Soc., 1999, v.43, N 1, p. 52-57, CA, 1999, V. 130, 252411
56. Choi Zel-Ho, W. Tyrra, A. Adam, Z. Anorg. Allg. Chem., 1999, v. 625, N 8, s. 1287-1292, CA, 1999, v. 131, 337058
Fluorine Notes, 2001, 14, 3-4
