Fluorine Notes, 2000, 10, 3-4
Use of perfluoroacylfluorides for synthesis of perfluoroalkylvinyl ethers. Perfluorinated vinyl ethers of a general formula CF3O(CF2CF2O)mCF=CF2A.E.Ershov, L.M.Popova Synthesis of relatively high-molecular perfluoropolyoxaalkylvinyl ethers is of a particular interest for creation of frost-resisting polymers. For synthesis of vinyl ethers (VE) both thermal decomposition of salts of the appropriate perfluoro-substituted carbonic acids and thermal interaction of acylfluorides of the same acids with solid oxygen-containing salts of alkaline metals in the presence of a solvent or without it are widely used. With the purpose of search of new monomers to produce fluoropolymers with a reduced glass-transition temperature two series of perfluoro-containing polyoxaalkylvinyl ethers have been synthesized: CF3O(CF2CF2O)mCF=CF2 (I) and CF3O(CF2O)nCF2CF2OCF=CF2 (II), where m, n = 1,2,3... . Oligomeric products of tetrafluoroethylene oxide addition to carbonylfluoride of the general formula CF3O(CF2CF2O)nCF2CFOF (III) were used as the starting material. VE (I) were synthesized by thermal decomposition of the reaction products of hexafluoropropene oxide (HFPO) with acylfluorides.
where m=n+1 The yield of IV was 55-96%. The mole ratio of the reagents, character of the catalyst (MF) and quantity of the solvent influenced significantly the yield while changes in the ratio of III/MF from 1 to 5 and in temperature from 20 to 30oC did not influence much the yield. It should be noted that in a number of the examples known in literature, a great excess of the solvent [1,2] was used and mainly CsF was used as a catalyst. Under such conditions, the yields of the goal products (products of HFPO mono-addition) usually did not exceed 60%. We have found that the reduction in the solvent amount to the ratio of CsF/diglyme brings to the increase of the product IV yield to 75%. Further replacement of CsF for KF allowed to increase the yield to 95%. The influence of the quantity of the used solvent may be explained by the fact that the product is partially soluble in diglyme and remains in the latter ( it is especially noticeably in an unjustified great amount of the diglyme) in separation of the organofluoric layer from the solvent followed by mechanical losses. The replacement of CsF with KF increases the yield of the goal product obviously due to the fact that KF is less active catalyst of the reaction of oligomeric addition of HFPO to acylfluorides [3], so when KF is used a probability of formation of by-products of addition of two and more HFPO molecules is reduced. Taken into account all abovementioned, in this study we used KF as the catalyst and carried out the synthesis at a definitely fixed molar ratio of the reagents, catalyst and solvent. Under these conditions, the yield of the goal product of mono-addition of IV attained 90-95%. The structure and purity of the compound of general formula IV was proved by GLC, IR and 19F NMR spectroscopy methods. An absorption band of carbonyl group of perfluoroacylfluorides was present in the IR-spectra at 1890cm-1. The 19F NMR spectra for all the products showed a signal of 129 ppm characteristic for the fluorine atom at the tertiary carbon atom. The molecular mass of compound IV was determined by titration with 0.1N solution of KOH. The products of mono-addition of HFPO to acylfluorides with general formula IV were used for the synthesis of VE I. With this purpose, either compounds IV in the presence of soda or their salts V in 10% aqueous solution of NaOH were subjected to thermal decomposition. F3O(CF2CF2O)mCF(CF3)CFCOF
+ 2NaOH 1.Thermal decomposition of sodium salt of perfluoropolyoxacarbonic acid. The salts of general formula V preliminarily carefully dried under IR- lamp and then in vacuum at 100oC were pyrolyzed at a temperature of 170-200oC. Unexpectedly, the pyrolysis of theses salts in addition to VE I gave a significant amount of acylfluoride IV and some amount of acylfluorides III: where m=n+1 The composition and structure of each of the reaction products proved by the data of GLC, IR and 19F NMR spectroscopy. The IR spectra of VE I showed a weak band of valence vibrations of the C=C bond at the oxygen atom at 1845cm-1. An intensive band of the carbonyl group was present in the spectra of acylfluorides at 1890 cm-1. It should be noted that in numerous examples described in patent literature about the synthesis of VE by thermal decomposition of a salt of RfOCF(CF3)COONa type no records of the formation of the by-products was found. For example, in studies [4,5] the appropriate vinyl ether was produced in a 90% yield by pyrolysis of C3F7O[CF(CF3)CF2O]2COONa. Any by-products were not isolated and identified. But in the thermal decomposition of the same salt in that study the formation of by- product CF3CF2CF2O[CF(CF3)CF2O]2CF(CF3)COF was found in 20% yield. The author of study [6] has determined the formation of acylfluorides only at pyrolysys of salts of the series of linear acids: RfOCF2CF2COOM The author of [6] assumes that in the pyrolysis of the linear salt of general formula RfOCF2CF2COONa, the appropriate acylfluorides are formed by the reaction of carbonylfluoride (eliminating at partial destruction of the salt) with vinyl ether, another pyrolysis product: RfOCF=CF2 + COF2 But this interaction is improbable, because carbonylfluoride is known to react with fluoroolefins only in the presence of polar aprotic solvents and special catalysts (CsF,KF) [7]. With the purpose to clarify the mechanism of the formation of acylfluorides in the synthesis of VE by thermal decomposition of salts of perfluoro-containing carbonic acids, the products of pyrolysis of linear salt CF3O(CF2))6CF2COONa were studied. Due to the specificity of the structure of the latter, its thermal decomposition can not bring to the formation of olefine and subsequently the olefine can not be converted to acylfluoride by the reaction with carbonylfluoride as it was assumed in paper[7]. The pyrolysis of the mentioned salt brought to the formation of acylfluoride CF3O(CF2O)6CF2COF in a yield of about 80% which was the sole liquid reaction product. By means of IR-spectroscopy, carbonylfluoride was found. In this case the formation of acylfluoride may be explained by interaction of carbonylfluoride with the starting sodium salt. CF3O(CF2O)6CF2COONa + COF2 It is known, that this reaction occurs very easily at high temperatures. Obviously, according to the same scheme ( including partial destruction of the polyether chain with carbonylfluoride elimination) the formation of side acylfluorides IV (in a 20-30% yield) takes place in the synthesis of VE by thermal decomposition of salts V. Perfluoropolyethers stabilized by treatment with fluorine and having no end functional groups are decomposed with the formation of carbonylfluoride usually at higher temperatures, about 400oC [8], but the presence of some catalysts ( for example, Al2O3 and oxides of other metals forming on the internal surface of a reactor) drastically reduces the destruction temperature [3,9]. A number of such oxides is part of the glass composition which was used for the pyrolysis reactor manufacture. But we obtained the same results when used reactors of glass and copper for thermal decomposition of salts of perfluoropolyoxacarbonic acids. Therefore the reduction in the destruction temperature of the polyether chain in the pyrolysis of salts in comparison with the destruction temperature of the stabilized PFPE should be explained most likely not by the influence of the reactor surface but by the presence of the end functional group. In this connection a known scheme of the formation of olefins [10,11] should obviously be completed with a number of reactions of PFPE chain destruction bringing to the formation of by-products of the pyrolysis of the salts of perfluorosubstituted carbonic acids:
Interaction of carbonylfluoride eliminationg according to the proposed scheme with starting salt V leads to the formation of side acylfluoride IV: CF3O(CF2CF2O)mCF(CF3)COONa
(V) + COF2 The formation of significant amount of acylfluoride IV in the pyrolysis of salts V noticeably reduces the yield of the goal product, VE (I). Thus, the widely used ( according to the literature data) method of synthesis of vinyl ethers by pyrolysis of the salts of the appropriate acids obviously is not the best method for VE synthesis of general formula I. The yield of vinyl ethers (I) in decomposition of salts V can be increased by an addition a dry soda in the reactor (2 moles of Na2CO3 per 1 mole of salt V). But the use of soda does not always exclude formation of some amount of acylfluorides. Also one should bear in mind duration, labour intensiveness of producing and particularly of careful drying amorphous salts of V structure used for the synthesis of VE. 2. Thermal decomposition of acylfluorides in the presence of oxygen-containing salts. A thermal reaction of acylfluorides with solid oxygen-containing salts of alkaline metals at a temperature of 250-270oC urgently requires good contact of the reagents. In the absence of a solvent it is possible to provide satisfactory contact by conducting the interaction in a fluidized bed when the solid salt is in suspended state in a reactor vertically placed. In this case a method to synthsize VE by interaction of the appropriate acylfluorides with the salt in an aprotic polar solvent was studied. The presence of such a solvent allows to reduce the pyrolysis temperature to 120-140oC. The reduction of the pyrolysis temperature is of great significance because already at 170-200oC in the process of thermal decomposition of salt V a significant amount of side product III is formed in addition to vinyl ethers I. To avoid the formation of compounds containing hydrogen in the process of VE synthesis, it is necessary to use absolutely dry solvents. Diglyme was used as a solvent in pyrolysis of acylfluorides III. The process was carried out in two stages in a glass reactor equipped with a turbine-type mixer, a thermometer and a Liebih refrigerator. In the first stage , salt Y was produced by reaction of acylfluorides III with a suspension of soda in diglyme at a temperature about 60oC. CF3O(CF2CF2O)mCF(CF3)COF(III)
+ Na2CO3 After the addition of the last portion of acylfluoride, the rate of the salt formation became slower and the temperature spontaneously went down. For completeness of the salt V formation, the temperature in the reactor (60oC) was kept by means of external heating. In the second stage the formation of VE took place as a result of thermal decomposition of the salt at heating of the reactor contents up to 120-140oC. CF3O(CF2CF2O)mCF(CF3)COONa(V) The pyrolyzate contained 95-99% of VE (I) and 1-5% of 2-hydroperfluoropolyoxaalkane of general formula CF3O(CF2CF2O)mCHFCF3. The preliminary distillate ( solvent stripper) collected in the first stage in producing the salt contained a small amount of unreacted acylfluoride III. At the conversion of 95% the yield of the vinyl ethers was 95%. Thus, the satisfactory results have been obtained in the synthesis of VE (I) by thermal decomposition of acylfluorides III in the presence of the solvent . The sole hydrogen-containing side product in a small amount (1-5%) was obtained under soft conditions of the pyrolysis (120-140oC). The possibility of formation of such a kind of compounds in the presence of water traces was obviously one of the reasons why this method was used relatively seldom. Experimental The IR spectra were recorded on a IKS-29 instrument. The 19F NMR spectra were measured at a frequency of 84.67MHz on a Brucker Spectrospin HX-90 instrument with the use of hexafluorobenzene as internal standard. 2-Trifluoromethyl-3,6,9-trioxaperfluorodecanoylfluoride (IV) (m=2). 300g of acylfluoride III(n=1) was added dropwise to 29g and 100ml of diglyme at vigorous stirring at 10oC. Then under the same conditions 157 g of HFPO was added during 20 minutes. The reaction products were distilled at 50 and 250oC. 450g of the mixture was produced containing 94% of product IV (m=2) and 6% of unreacted III (n=1) according to the GLC data . By rectification 424g of IV (n=1) was isolated: b.p. 101.5oC, 91% yield. 19F NMR (CFCL3): 27.3 (1 F, COF), 55.6 (t, J=10Hz, 3F, CF3), 81.5 (m, 3F,CF3)83.7; 90.8(m,2F,FCF0, 87.5(m,8FCF2), 89.3 (m,2F,CF2), 89.6(m,2F,CF2) and 128.8(dd,J=19Hz, IF,CF). The rest compounds of general formula IV for m=1 (b.p.61oC), m=3(b.p.137oC) and m=4(b.p.-58oC/9 Torr) were produced similarly. 2-Trifluoromethyl-3,6,9-trioxaperfluorodecanoate of sodium V (m=2) was produced by interaction of 420g of acylfluoride IV (m=2) with 720 mL of 10% aqueous solution of NaOH at 0oC. Water from the produced salt solution was evaporated under IR-lamp at a temperature above 100oC, the salt was then dried under vacuum.476 g of an equimolar mixture of salt V(m=2) and NaF was produced. The rest salts of general formula V (m=1,3,4) were produced similarly. Thermal decomposition of salt V (m=2) 60g of Na2CO3 was added to 476g of the equimolar mixture
if salt V(m=2) and NaF and heated at 170-200oC. 360g of pyralizate
was produced which contained 66% of 3,6,9-trioxaperfluorodecene (I)(m=2),
26% of acylfluoride IV(m=2), 6% of acylfluoride III (n=1) and 2% of high-boiling
unidentified compound according to the data of GLC. The reaction mass was
subjected to rectification. VE I (m=2) yield was 221g (61%)(b.p. 89oC).
19F NMR (CFCl3): The thermal decomposition of the rest salts of general formula V resulted in the goal VE of general formula (I): for m=1 b.p.=46oC, m=3 b.p.=121-122oC and for m=4 b.p.=160oC Synthesis of 3,6,9-trioxaperfluorodecene-1 (I) by interaction of acylfluoride IV (m=2) with soda in the presence of diglyme. 420g of acylfluoride IV(m=2) was added dropwise to 170g of Na2CO3 and 85 mL of diglyme (abs.) at vigorous stirring at 60oC. The reaction mass was then heated at 120-140oC, after the reaction completion 346g of pyrolyzate was produced which contained 98.5% of the goal VE (I) (m=2) and 1.5% of 2-hydro-3,6,9-trioxaperfluorodecane according to the data of GLC, IR- and 19F NMR spectroscopy. The conversion of acylfluoride IV was 97.8%, 19F NMR (CFCl3): 55.2 (t,J=10Hz,3F,CF3), 87.3(m,8F,CF2),89.3(m,2F,CF2),113.9(1F,=CF2),121.6(1F,=CF2) and 134 (1F,CF) The pyrolysis of acylfluorides of general formula IV resulted in VE of general formula I with m=1,3,4 in approximately the same yield. REFERENCES
|
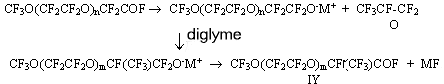
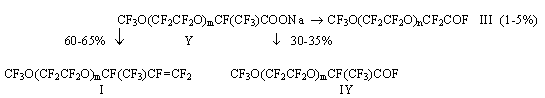
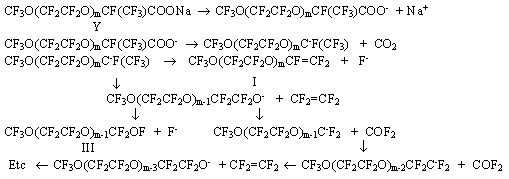
Fluorine Notes, 2000, 10, 3-4
