Fluorine Notes, 2000, 9, 7-8
S.E.Gonek, D.Ya. Kasaeva N,N-Dimethyl-N'-perfluoroalkylamidopropylamines (AAPFKK) (1) are intermediate products for synthesis of quarternary ammonium salts used as a main component for high-performance compositions of film binders and foam formers [1,2], corrosion inhibitors, emulsifiers, dispersants and also surfactants operating in anhydrous media [3,4]. CF3(CF2)nCOOC2H5 + NH2(CH2)3N(CH3)2 n = 5 (1a); n = 7 (1b). Methods to produce N,N-dimethyl-N'-perfluoroalkylamidopropylamines from esters of perfluorocarbonic acids and N,N-dimethylpropanediamine at a ratio of components of 1:1,5 in an inert solvent or without it at a temperature of 40oC maximum are known. Thus, according to [1], N,N-dimethyl-N'-perfluoroctylamidopropylamines were made from butyl ester of perfluorooctanic acid and N,N-dimethylpropanediamine in equimolar ratio at 23-40oC for 7 hours. N-butanol formed as a result of the reaction was distilled at a reduced pressure. The yield of the goal product attained 92%. The necessity of vacuum distillation of n-butanol and the presence of wastes are disadvantages of the given method for industrial use . According to [2], N,N-dimethyl-N'-perfluoro-caprylamidopropylamine was made by interaction of ethyl ester of perfluorocaprylic acid and N,N-dimethylpropanediamine taken at equimolar ratio in a medium of diethyl ether at a temperature below 15oC. To isolate the goal product, vacuum distillation was used, the yield was 91%. The following should be mentioned as disadvantages of the mentioned method: the use of solvent, fractional distillation of the solvent and of the alcohol forming in vacuum and also the presence of wastes. The common shortcoming of listed methods [1,2] is multistage synthesis, because the starting esters were produced either from perfluorocarbonic acids and alcohols or from acid halides of the appropriate perfluorocarbonic acids and alcohols. Methods to produce N,N-dimethyl-N'-perfluoroalkylamidopropylamines from perfluoroacylfluorides and N,N-dimethylpropyldiamine (DMPD) are also known. A method to produce (1) directly from perfluoroacylfluorides (2) and DMPD was developed earlier at RSC Applied Chemistry. Acid fluorides of perfluoroheptanoic (perfluoroenanthic) and perfluorononanoic (perfluoropelargonic) acids were chosen as the starting perfluoroacylfluorides. The reaction was carried out in difluorodichloroethane (R-113) at 40oC for 2 hours followed by neutralization of hydrofluoric salt of AAPFKK (4) forming during the process with dry soda according to the scheme: After the neutralization, the reaction mixture was filtered and the solvent (R-113) was distilled , as a result goal products (1) were produced in a yield below 80%. But the multistage process, the use of the expensive solvent and losses of the product during its separation make the mentioned method uneconomical. We have developed a more practically feasible single-stage wasteless method without solvent to produce AAPFKK (1) from perfluoroacylfluorides (2). The method is based on interaction of compounds (2) with DMPD (3) under conditions of an adiabatic process in the absence of solvent at an elevated temperature. The heat efficiency of the reaction of interaction of acid fluoride of perfluoropelargonic acid with N,N-dimethylpropanediamine was determined by a method of isothermal thermometry as a result of the analysis of a thermogram and was 121 kJ/mol [5]. It was sufficient for thermal decomposition of salt (4) on AAPFKK (1) and HF and for removal of the forming gaseous product from the reaction mixture. The process temperature was kept within the range of 90-120oC and controlled by the rate of DMPD (3) charge into the reaction zone. The yield of the goal products attained 98%. The choice of the temperature was caused by the fact that below 80oC a mixture of goal product (1) and its salt (4) was formed and at a temperature above 120oC a decrease in AAPFKK (1) yield was observed as a result of its resinification (see Table). Upon the process completion , the reaction gas was kept at 90oC in a flow of inert gas ( for example, nitrogen) till full decomposition of salt (4) with the purpose to remove HF completely. The control over the reaction was executed by determination of the fluoride-ion content in the goal products and by solubility of the latter in water ( AAPFKK is almost insoluble in water). The composition and structure of N,N-dimethyl-N'-perfluoroamidipropyl amines (1) synthesized by the mentioned method have been confirmed by elemental analysis, gas liquid chromatography, IR and proton NMR spectroscopy and coincide with the known values for compounds (1) obtained by different methods listed earlier. In the proton NMR spectra of compounds (1), resonance signals of protons of the methylene groups as three triplets with chemical shifts of 3.39, 1.6 and 2.38 ppm and a singlet of the methyl groups with a chemical shift at 2.16 ppm were observed, all of them corresponded to the assigned structures taken into account their integral intensities. The IR spectra of produced compounds (1) in high-frequency region have characteristic bands of absorption of NH group within a range of 3250-3340cm-1, bands of absorption of valence vibrations of the methyl and methylene group within a range of 2805-2970 cm-1, a band of absorption of valence vibrations of the carbonyl group at 1720 cm-1, deformation vibrations of NH at 1500 cm-1 and intensive bands corresponding to valence vibrations of CF in a range of 1000-1300cm-1. Experimental The IR spectra were obtained on a IKS-29 instrument ( film or suspension in liquid paraffin). The proton NMR spectra of solutions of the substances in DMCO-d6 were recorded on a RIA -2309 spectrometer (90MHz) with GMDS as an internal standard. N,N-Dimethyl-N'-perfluoroheptylamidopropyl amine (1a).20 L of tridecafluoroheptanoyl fluoride was charged into a 60L reactor equipped with a stirrer and a bubbler to charge inert gas from a batcher , the product was heated to 70oC, purged with inert gas and N,N-dimethylpropanediamine was added, 11.6 L in total. The feeding rate was regulated according to the reactor temperature and was chosen in such a way that the temperature would be 95+5oC. After completion of DMPD feeding, the reaction mixture was kept at 95oC for another 2 hours, then the product was discharged. The yield of compound 1a was 98%, resinification degree was 0.9%. Table. Influence of the reaction temperature on yields of N,N-dimethyl-N'-perfluoroalkylamidopropylamines (1)An improved method to produce N,N-dimethyl-N'-perfluoroalkylamidopropylamines
![]() CF3(CF2)nCONH(CH2)3N(CH3)2 (1à,b)
CF3(CF2)nCONH(CH2)3N(CH3)2 (1à,b)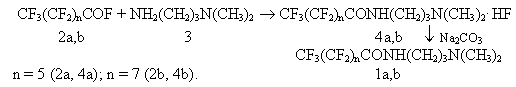
| No | F-anhydride | Quantity of F-anhydride | DMPD quantity,L | Reaction temp.,oC | Yield, % | Resinification % |
1 | 2a | 20 | 11.6 | 95 | 98.6 | 0.9 |
2 | 2a | 18 | 10.5 | 82 | 98.7 | 0.6 |
3 | 2b | 18 | 8.9 | 114 | 98.0 | 0.8 |
4 | 2b | 19 | 9.4 | 120 | 97.9 | 1.0 |
N,N-Dimethyl-N'-perfluorononylamidopropylamine (1b) 40.8 g (0.44 mol) of N,N-dimethylpropanediamine was added dropwise to 186.4 g (0.44 mol) of perfluorononanoyl fluoride at stirring and a temperature below 70oC in an inert gas flow . The feeding rate of DMPD was chosen in such a way that the reaction mixture temperature would be 90+2oC. After the completion of DMPD feeding, the reaction mass was kept at 90oC for 2 hours, then the product was discharged. The yield of compound 1b was 214.8g (98%), light yellow crystals, melting point of 24oC. Found,%: C 30.60; H 2.70; N 5.60; F 58.2. Calculated, %: C 30.60; H 2.37;N 5.10; F 59.94.
The control over the reaction was executed by determination of the amount of fluoride-ion in compound 1b, the process was finished when the content of F-ion reached a value of 0.0013g/g of the product at which the goal product was insoluble in water.
Thus, as a result of the conducted investigations, a method to produce N,N-dimethyl-N'-perfluoroalkylamidopropylamines by interaction of industrial perfluoroacylfluorides with N,N-dimethylpropanediamine has been developed. It differs from the known ones for the following features: the reaction was carried out in the absence of solvent, in single-stage process and at a temperature of 80-120oC with the purpose to increase the yield and simplify the process.
References
1.USA patent No 3350218
2. USA patent No 2764602
3. New ideas in technology of fluorine compounds/ edited by N. Isikawa- M.: Mir, 1984. p.385-422
4. Fluorine compounds. Synthesis and application/ edited by N. Isikawa M.: Mir, 1990. p.157-182
Fluorine Notes, 2000, 9, 7-8
