Methods for hexafluoroacetone production
(1,1,1,3,3,3-hexafluoropropanone-2)
T.E.Fiodorova
Introduction
For the first time hexafluoroacetone (HFA) was produced in 1941 by N.Fukuhara
and L.A.Bigelow by direct fluorination of acetone. HFA is an extremely toxic
reactive compound with the following main physical characteristics:
-
Molecular mass 166.03 (1)
-
Boiling point (760 mm of mercury column) 27.5oC
-
Melting point -125.45 -129oC (1)
Nonflammable HFA is an active electrophile and reacts with alkenes, ketenes,
aromatic compounds, compounds containing active hydrogen atom etc. HFA does
not form homopolymers but is readily copolymerized with various monomers
to form chemically and heat resistant polymers. A field of HFA application
is wide enough: production of solvents ( hexafluoroiso-propanol, for example),
pharmaceuticals and agrochemicals, anticorrosive coatings, emulsifiers, dyes
etc.
1.HFA production from acetone
and its derivatives
1.1. HFA production by
direct fluorination of acetone
In 1941 N. Fukuhara and L.A.Bigelow reported about a process of gas-phase fluorination
of acetone by fluorine-nitrogen mixture at different ratios of F2:CH3COCH3:N2.
According to the authors, it was easy to fluorinate acetone in a reactor packed
with copper gauze. The reaction products were separated and identified. The
main reaction products were: HFA, monofluoroacetone, trifluoroacetyl fluoride,
oxalyl fluoride, carbonyl fluoride and tetrafluoromethane. The fraction (b.p.
-35- -16oC) containing HFA was separated in amount of 5-10% of
the whole liquid volume (4).
The reaction of direct fluorination is dangerous and difficult to control, therefore
this method was not spread widely but it is still of interest.
An attempt to use AgF (64) as a fluorinating agent for acetone fluorination should
be mentioned, but it also did not find further development.
In the late 70-s, gas phase fluorination of acetone was carried out in a fused
alumina reactor, a steel tube with inside alumina porous member fused as
a tube. The fluorine-nitrogen mixture was fed into the tube of alumina and
the compound to be fluorinated was fed into the intertubular space. The maximum
temperature was 148oC, the HFA yield was 38-43%. Fluorinated acetones
( from mono- to pentafluoroacetone) and a number of other products were produced
as by-products (5).
The use of the low temperature gradient method in the presence of NaF at -100oC
resulted in 39% HFA yield. According to the authors, sodium fluoride provided
more gentle fluorination conditions and adsorbed the evolved hydrogen fluoride
to form sodium bifluoride. Trifluoroacetylfluoride and octafluoropropane
were formed together with HFA. In the authors opinion, the process parameters
are subject to optimization and the process can be brought to industrial
implementation.
1.2. HFA production from
perhalogenacetones.
1.2.1.Catalytic methods
Fluorination with anhydrous hydrogen
fluoride
The halogen exchange reaction between hexafluoroacetone or chloroacetones and
anhydrous hydrogen fluoride in the presence of a catalyst containing chromium
(III) salts is the basis of the industrial method to produce HFA (1,6).
In the early 60-s, Farbwerke, Du Pont and Allied Chem. Companies developed catalytic
methods for gas phase fluorination of a number of compounds, including perhalogenacetones,
in the presence of a catalyst based on Cr(III) (7,8,9). The approach described
in a Hoechst AG patent used chromium oxyfluorides produced by interaction
of Cr(OH)3 (or "Guignet Green" commercial pigment) with hydrogen
fluoride diluted with inert gas (nitrogen, argon) at a ratio of 1:10 at a
temperature of 350-400oC. The atomic ratio of the elements in
the catalyst produced was Cr:F:O=1: (1.5-2): (1-1.5).
Hexachloroacetone was fluorinated on the catalyst produced at 250oC
to yield HFA in 81% as per hexachloroacetone (0.55M) and 4.02M HCl.
In works (8,9) CrO3 was used as a catalyst. It can be produced by
Cr2O3*2H2O dehydration (8) or CrO3 reduction in an aqueous solution to form a gel-like product with following
drying (10).
According to (8), HFA was produced in a tubular reactor of 3.5 m length and 5.1
cm diameter filled with 5 kg of Cr2O3 gel. As a result
of the reaction, HFA, chloropentafluoroacetone and dichlorotetrafluoroacetone
were produced at hexafluoroacetone conversion of 98%.
Paper (9) describes fluorination of perhalogenacetones over Cr2O3 catalyst produced by precipitation of chromoxide hydrate from a
water-soluble chromium salt (nitrate, for example) with an alkali reagent
followed by filtration, washing, drying and pressing into tablets. The tablets
produced were preheated in a reactor at a temperature of 370 - 400oC.
HFA was produced by fluorination of dichlorotetrafluoroacetone at a temperature
of 330-380oC. The yield attained 21%, the rest was unreacted dichlorotetrafluoroacetone.
In the early 70-s, a continuous process to fluorinate perchloroacetone was developed
using three-zone catalytic bed of chromium (III) hydroxide treated with hydrogen
fluoride(11). The temperatures of every bed were different: 234-236oC,
260-271oC, 280-340oC respectively. The contact time
in every bed was different also: 6-10.9 s, 3.8-4.8 s and 3.5-6.2 s. Every
catalyst bed was separated with a bed of aluminum oxide. As a result, a reaction
mixture containing HFA, CF3COF,CF3CHO and CF3CF2COCF3 was produced in 95.3-97.4% yield at the total fluorination degree of 97.2-98.9%.
Work (12) proposed a mixture of oxalates of Cr (III) and Ni(II) as their hydrates
, Cr2(C2O4)3*6H2O
and NiC2O4*2H2O, as fluorination catalyst.
A catalyst with the specific surface of 360m2/g was treated with hydrogen
fluoride in a mixture with nitrogen at a ratio of 1:4.
In the 70-80s., some works were carried out with the purpose to increase the
process selectivity and the catalyst life time without reduction in its activity.
It was found that chromium oxide catalysts produced by reduction of CrO3 of the " Guignet Green" pigment , precipitation of trihydroxide from aqueous
solutions of Cr (III) salts possess  -CROOH orthorhombic crystallinity effect. Treatment
of such a gel-like catalyst with water at 250oC for 20h leads
to its structure transition to Cr (III) oxide hydroxide (CrOOH) hexagonal
structure. CrOOH was applied on calcium difluoride. The life time of this
catalyst in the hexachloroacetone fluorination was 700h, the life time of
the catalyst with -orthorhombic structure was 400h (13). -CROOH orthorhombic crystallinity effect. Treatment
of such a gel-like catalyst with water at 250oC for 20h leads
to its structure transition to Cr (III) oxide hydroxide (CrOOH) hexagonal
structure. CrOOH was applied on calcium difluoride. The life time of this
catalyst in the hexachloroacetone fluorination was 700h, the life time of
the catalyst with -orthorhombic structure was 400h (13).
Work (14) proposed Cr2O3 catalyst as microspheres of 0.1-0.3mm
diameter according to sol-sel technology. The use of this catalyst
in the hexachloroacetone fluorination resulted in the following reaction
products: HFA (75%), chloropentafluoroacetone, small amounts of dichlorotetrafluoro-acetone
and trichlorotrifluoroacetone at 76% hydrogen fluoride conversion.
Another method to produce a long-lived catalyst is described in (15). Cr(OH)3 without ionic admixtures was dried and calcined at 450-550oC.
Paper (16) considers a procedure to produce Cr2O3 by CrO3 reduction with methanol followed by activation of the Cr2O3 produced with
hydrogen fluoride and trifluorotrichloroethane at 400oC.
Disproportionation of perhalogenacetones.
HFA and chloropentafluoroacetone are produced by catalytic disproportionation
of perhalogenacetones in the presence of Cr2O3 in gas
phase at 225-400oC according to the scheme:
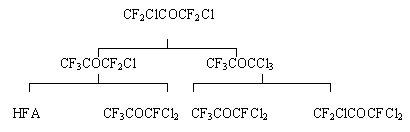
The yield of HFA from dichlorotetrafluoroacetone is 12.2% and 40.7% from chloropentafluoroacetone.
The authors believe that the advantage of this route is that the mixture
of HCl and HF difficult to separate is absent, while it is formed in the
fluorination with hydrogen fluoride.
1.2.2. Non-catalytic methods
According to the authors, a simple and controlled method to produce HFA by fluorination
of perhalogenketones is their interaction with KF taken in a stoichiometric
amount and suspended in tetrahydrothiophene-1,2-dioxide, an inert polar solvent.
HFA is produced from dichlorotetrafluoroacetone in 67% yield at a temperature
of 180-190oC at the starting perhalogenacetone conversion of 56%.
It is possible to use other fluorides: CsF, LiF, AgF but potassium fluoride
is the most preferable (18).
The study of the properties of polyfluoroacetonylfluorosulfates has found that
their interaction with SbF5 at a temperature of 35
њ
C results in the formation of HFA in yield of 77.5%yield (19).
2. Hexafluoroacetone production by
oxidation of hexafluorothioacetone (HFTA) dimer
In the 80-s a method to produce HFA from 2,2,4,4-tetrakis(trifluoromethyl)-1,3-dithioethane,being
hexafluorothioacetone (HFTA) dimer, and also to produce the dimer itself
was developed (2,20-24). HFA was produced by treatment of the dimer with
oxidizer in a medium of aprotic solvent in the presence of alkali metal fluoride
as a catalyst at a temperature of 140-149
њ
C in 80-89%yield. The process takes place according to the scheme:
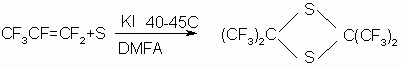
The following substances were used as oxidizers: HgO,Ag2O,Cu2O,CuO,MeIO3,
MeIO4 where Me is alkali or alkali-earth metal (21), also air,O2,O3,NO2,NO
(22). Dimethylformamide, dimethylsulfoxide and N-methylpyrrolidone were used
as aprotic solvents.
The dimer can be converted to HFA for 24 h without oxidizer fed in 25% yield.
The dimer was also used , beside HFA production, to produce perfluoroisobutene
by its treatment in an aprotic solvent with fomaldehyde or with a compound
forming formaldehyde ( paraformaldehyde for example) in the presence of alkali
metal fluoride at 110-150
њ
C(24).
The authors considered the following as the advantage:
-
the proposed method to produce the dimer is easier than that used for the
dimer production earlier according to the reaction of bis(perfluoroiso-propyl)mercury
with sulfur vapors at 445oC and the yield of HFTA of 60% followed
by its dimerization in the presence of excess triphenylphosphine (25).
-
In comparison with the known methods to produce HFA (hexafluoro-propylene
epoxidation followed by isomerization to HFA, high-temperature halogen
exchange with hydrogen fluoride/Cr(III), perfluoroisobutene oxidation)
, this method is cheaper, less labor-intensive and is carried out in
standard equipment (2).
3. Hexafluoroacetone production by
oxidation of olefins
3.1.Hexafluoropropylene
oxidation
A study of the hexafluoropropylene oxidation reaction in a flow-type reactor
has found that fluorides such as CsF,RbF,LiF,KF and NaF catalyze this reaction.
A multi-component mixture containing CF3COF, COF2,
CF3CF2COF, HFA, CF3OCF2COF etc.
has been produced. In addition to oxygen, CrO3 and KMnO4 may be used as an oxidizer. Works carried out in Russia have shown that chromium
(VI) oxide reacts with HFP to form carbonyl compounds. The reaction was carried
out in an autoclave at stirring for 3 days. The starting components were
HFP,HF,CrO3. The synthesis products were run through water, the
water solution was then extracted with ether. There were produced 24% HFA
and 23% of ethyl ether of trifluoroacetic acid (27).
An investigation of the HFP oxidation in HFA in details has been performed by
Daikin, Japan company. A high yield of HFA was attained by oxidation with
oxygen over Al2O3 treated with SF4 at 350oC
or with HF at 450oC (28). In addition to HF and SF4,
in paper (29) other promoters are mentioned: SiF4, SF6,
NH4F, organic fluorides including freons. HFA was produced in
a yield of 4 to 21.2% (mol) at a temperature of 170oC and pressure
of 5kg/cm2. Further a blend catalyst, fluorinated Al2O3 -SiO2, was used. The oxidation was carried out both in the presence
of water and without it. The process over fluorinated aluminum oxide ( without
SiO2) was conducted in the presence of water. A mixture of hexafluoropropylene
with oxygen containing water in amount of 0.008-0.0016mol/HFP mol was passed
over the fluorinated Al2O3 at 175oC. The
conversion was 15.4%, selectivity 70.4% after 450 h. If the process was carried
out in the absence of water, the conversion of HFP was reduced to 34.4% but
the selectivity was reduced to 29.8% after 160 h (30).
Besides the catalyst based on aluminum, other ones were studied also. Paper (31)
has considered the HFP oxidation process over oxides of Sn (IV), Fe ( III),
In (III) at a temperature of 150-300oC. Using SnO2 catalyst and at 250oC, the conversion of HFP was 70.6% the selectivity
to HFA 53.9%. Under the same conditions and Fe2O3 catalyst,
the figures were 49.3% and 52.4% respectively, in case of using In2O3:
54.1% and 52.2%. CF4 and COF2 were formed together
with HFA. Comparative experiments with V2O5 and PbO2 showed their low activity. Also, commercially produced metals of the platinum
group, applied on activated carbon, have been investigated. The HFP oxidation
was carried out in a circulation reactor at 110-190oC. At the
same time the process was carried out using oxides of vanadium, molybdenum
and silver at 250
њ
C. The highest HFP conversion of 63.2(mol)% was reached with a 5% Ru/C catalyst
at HFA selectivity of 87.4% and 120
њ
C. The use of a 0.5% Pd/C catalyst brought to 54.7% conversion and 87.4% (mol)
selectivity at 150oC. The authors of these investigations considered
the HFP oxidation the most simple and convenient method to produce HFA.
3.2. Oxidation of
perhalogenisobutenes
Oxidation of perhalogenisobutenes is one of the first directions in the development
of HFA production methods. A.L.Henne, J.W.Shepard, J.Evan proposed their
method as an alternative to the direct fluorination. Potassium permanganate
directly oxidized (CF3)2C=CCl2 which was
produced in its turn by a multi-stage method from CF3C(CH3)ClCH2Cl.
In spite of the multi-stage mode, the authors considered the use of commercial
compounds and common operations as the advantage. The yield in every stage
attained 90-95% taken into account recycle. HFA yielded in 60% (33). First,
perfluoroisobutene oxidation with an aqueous solution of potassium permanganate
at 100oC resulted in the formation of HFA hydrate which was then
converted to HFA according to the Henne procedure by the hydrate addition
to phosphorus (V) oxide dropwise and heating to 30oC. The yield
of HFA attained 27%, perfluoroisobutene conversion was 67% (34).
The yield of HFA has been found low (~16%) in case of oxidation of fluorine-hydrogen
containing isobutenes with potassium permanganate because this compound does
not contain the halogen atom joined directly to the olefin carbon. Replacement
of one hydrogen with bromine followed by oxidation with permanganate increased
HFA yield to 55% (35).
Later, oxygen was proposed as an oxidizer(36). The process was carried out in
a nickel tube at a temperature of 500oC. HFA yield was 96% of
the theoretical one. The technical product contained admixtures of perfluorocyclobutane
and butenes. Oxidation of another derivative of perfluoroisobutene, (CF3)2C=CFOR
(where R=alkyl) at photoinitiation with a mercury lamp led to 19.2% yield
of HFA (37).
Hydrogen peroxide in the presence of carbonates or hydroxides of alkali or alkali-earth
metals may be used as an oxidizer. Perfluoroisobutene was added to 30% hydrogen
peroxide and methanol, the mixture was then treated with aqueous potassium
carbonate. The both stages were carried out at room temperature. HFA yield
as hydrate was 40.8% (38).
4. HFA production by isomerization
of hexafluoropropylene oxide (HFPO)
4.1. Isomerization in the
presence of fluorides
Antimony pentafluoride was used for HFPO isomerization to HFA (39) in the early
60-s, in the 80-s the process used HFPO contact with antimony pentafluoride
at a temperature of 31oC and a pressure of 0.3-13.76 MPa. HFPO
conversion was 99% (40).
The following other fluorides were used:
-
hydrogen fluoride in equal mass parts with HFPO. In this case HF was both
the catalyst and the solvent. By this method HFA was produced in over
90% yield at 100oC. At HFPO conversion of 95.7% HFA was produced
in 98.3% selectivity, at 99.1% HFPO conversion Selectivity of HFA was
94.3% (41).
-
Cobalt trifluoride at a temperature of 40-90oC, the yield was
99.2% at complete HFPO conversion (42).
4.2. Isomerization in the
presence of the Lewis acids
In the early 60-s a gas phase process to produce HFA by HFPO isomerization on
Al2O3 catalyst as spheres of 3 mm diameter on a support,
6 mm glass rings, was developed. The process temperature was 100oC,
nitrogen was used as a gas-carrier. The reaction was carried out to 49% yield
of HFA. At the same time a liquid-phase method at room temperature in liquid
SO2 in the presence of AlCl3 as a catalyst was developed.
HFA was produced in 79% yield (43).
But a low yield of HFA required further investigations on search of more effective
catalyst.
Work (44) describes a process with the use of fluorinated alumina or silica-alumina
at 170oC. The use of alumina containing 10% of fluorine resulted
in HFPO conversion close to the calculated one. Catalyst regeneration was
performed with oxygen.
To avoid a reduction in catalyst activity, it was proposed to carry out the isomerization
in the oxygen presence (45).
Al2O3 promotion with fluorine-oxygen-containing compounds
such as perfluoroethylisopropylketone, perfluoroethylhexylketone, HFPO trimer
increases the yield to 93.5-97.4% at 100% HFPO conversion and at a process
temperature of 80-120oC. Tetrafluoroethylene and carbonyl fluoride
were by-products (46).
At 100% conversion , HFA yield increases to 99.5% if Al2O3 is promoted with antimony pentafluoride in amount of 0.03-0.3 equiv. blowing
dry nitrogen together with antimony pentafluoride vapors through calcined
aluminium oxide at a temperature of 5-100oC. The isomerization
process had a temperature of 25 to 220oC (3).
97% of HFA was attained at 100% HFPO conversion using fluorinated -Al2O3 containing 31% fluorine
(47).
I
n work (40) Al2O3 was promoted with 0.01-0.04 mol/g of
hexafluoro-propylene at 400-650oC. According to the authors, more
cheap and simple in preparation is a catalyst promoted with hexafluoropropylene
oxide. The yield of HFA reached 99% at HFPO conversion of 100% (49).
94% HFA yield at 98% HFPO conversion was attained using Cr2O3 treated with hydrogen fluoride in a nitrogen flow at 150-250oC.
The fluorine content in the catalyst reached 13% (50).
5. Other methods to produce HFA.
5.1. HFA production from
carbonyl compounds and ethers.
HFA has been produced by electrochemical fluorination of CF3CO2CHCH3 to produce CF3CO2CF(CF3)2 followed
by its treatment with sodium fluoride to form HFA and CF3COF (51).
Investigation of reactions of perfluoroalkylbromides or iodides with derivatives
of carbonic acids in the presence of Zn powder has found that interaction
of trifluoroethylacetate with bromotrifluoromethane at 20oC in
pyridine led to the formation of HFA in 54% yield (52).
HFA and its hydrates have been produced from hepta- or octafluoroiso-butenyl
ethers or their derivatives by ozonation or thermal oxidation with oxygen
on an activated carbon catalyst. The produced compounds were decomposed with
water. Ozonation of heptafluoroisobutenylmethyl ether led to 69.7% yield
of HFA (53).
Decomposition of branched perfluoroalkyl ether fluid with anhydrous aluminum
chloride in an oxygen-free medium leads to the formation of volatile products,
mainly HFA and fluorine-chlorine-containing compounds and oligomers as well
(54).
5.2. HFA production from olefins
and its derivatives.
HFA together with perfluoropropylsulfide was produced from HFP and SOF2 in a
close system in a tubular reactor containing CsF at 120oC for
40 hours (55).
HFA in 77% yield was produced by heating a mixture of HFPO and perfluoroisobutene
for 10h at 170oC (56).
Nucleophilic oxidation of 3-chloropentafluoro-1-propene led to the formation
of 3-chloropentafluoropropene-1,2,-oxide which subjected to antimony pentafluoride
formed a mixture of fluorinated acetones in total yield of 81.1% at the ratio
of chloropentafluoroacetone:HFA=72:28 (57).
5.3. HFA production from HFCs.
One of the possible ways to produce HFA and other perfluoroketones is electrochemical
oxidation of aliphatic secondary and tertiary hydrofluorocarbons in a medium
of fluorosulfonic acid followed by pyrolysis of perfluoroalkylsulfates obtained
over cesium fluoride to the appropriate ketones (58).
In the middle 90-s a catalytic process of oxidation of hydrofluorocarbons including
1,1,1,3,3,3-hexafluoropropane with oxygen and fluorine in the presence of
Al2O3 catalyst at 50-300oC and the contact
time of 2-60s was developed (59).
5.4. Other methods to produce
HFA.
The Grignard reagents can be converted to ketones including HFA as well according
to the reaction:
CF3MgI + CF2CN -> HFA
to form HFA in 33% as per CF3MgI (60).
Diazomethane oxidation with molecular oxygen under exposure to radiation of 305nm
wave length brings to the formation of HFA-O-oxide which is converted to
HFA under exposure of 590nm wave length (61).
Work (62) has considered a way to produce HFA from its oximes by their heating
with concentrated mineral acid (H2SO4). Oximes were
produced from heptafluoroisobutyroyl carbanion alkylammonium salts. HFA yield
was 78%. More over, HFA is possible to produce from 1-methoxy-1-perfluoroisobutene
by its interaction with sodium nitrite in 59%yield and from methoxy-2H-octafluoro-isobutane
and N2O4 in 51% yield. Also, HFA is formed in bis(trifluoromethyl)-sulfine
photolysis (63).
Conclusions
Commercially important routes to produce HFA are
-
fluorination of acetone halides with hydrogen fluoride
-
hexafluoropropylene oxidation
-
hexafluoropropylene isomerization
All three methods are catalytic and the main problem in their development was
to establish an effective catalytic system, technology of separation of the
multi-component reaction mixture, purification and separation of the goal
products. The method of fluorination of acetone halides was implemented in
industrial production by American companies, Du Pont and Allied Chem Corp.
(Allied Signal at present), as early as in the 60s. By the 80-s no publications
on further improvement of this process were found.
The method of hexafluoropropylene oxidation was developing in the 70-80s in Japan
(Daikin Co Ltd) and considered as the most convenient and simple in the authors
opinion. Publications on the method came to the end by the late 80-s.
Development of the process of hexafluoropropylene oxide isomerization was conducted
in the USA (Du Pont and AlliedSignal), the USSR (Russia) and Japan (Asachi
Glass Co, Daikin Co Ltd) since the middle 60-s to the middle 80-s. The publication
of (42) is not an exception because the patent application was filed in 1980.
The publications of the last years are rather of a scientific interest but patent
(59) of Allied Signal possessing great experience in development of the technology
and production of HFA.
References
-
Kirk-Othmer. Encycl. Chem. Technol. 3 rd Ed. 1980, v.10, p.882
-
M. Van der Puy. L.G. Anello, Organic Syntheses, 1985, v. 63, p. 154-159
-
I. Vilenchik and G.Iakurnova .Pat. SU 764312, puplish 1995
-
JACS, 1941, v.62, p.788-791
-
Pat. EP 31519, 1980
-
W. Clark, R.Lagow, J. Fluorine Chem.,1991, 52(1), p 37-43
-
Pat. Fr. 1343392, 1963
-
Pat. Fr. 1372549, 1964, CA 1965, v. 62, 6397b
-
Pat Fr. 1369784, 1964; Pat Fr. 1369782, , Allied Chem. Corp., 1964
-
Pat. US 2271356, 1942, CA, 1942, v. 36, 3509'
-
Pat DE 2221844, 1972, CA 1973, v.78, 42856
-
Pat. DE 2221849, 1972, CA, 1973, v.78, 42858
-
Pat US 3978145, 1976
-
Pat EP 55652, 1981
-
L. Marangoni, G. Rasia. Chem .End., 1982, 64(3), p.135-140
-
Pat. EP 146464, 1985, CA, 1985, v.103, 123004
-
Pat. Fr. 1369785, 1964
-
Pat US 3379765, 1968
-
A.V.Fokin, A.I. Rapkin, I.N. Krotovich, Izvestia Akad. Nauk SSSR, "Khimia",
1987 (6), p.1368
-
L.G. Anello, M. Van der Puy, J. Org. Chem., 1982, 47(2), p.377-378
-
Pat US 4334099, 1982, CA, 1982, v.97, 109555
-
Pat US 4337361, 1982, CA, 1982, v.97, 71947
-
Pat US 4337362, 1982, CA, 1982, v.97, 71948; Pat EP 66678
-
Pat EP 68080, 1983,CA, 1983, v.98, 161296
-
W. Middleton, E.G. Howard, W.H. Sharkey, J.Org. Chem, 1965, 30, p.1375
-
J.A. Wethington, T.A. Gens, W.H. Christie, Proc. UN Intern. Conf. Peaceful
uses At Energy. 2 nd, Geneva, 1958, v.20, 132. CA, 1960, v.54, 23674e
-
G.G. Belenky, L.S German, I.L. Knuniants. Izv. Akad. Nauk SSSR, Khimia, 1968,
(7), p.1631
 -
Pat DE 2624349, Pat US 4057584, 1977
-
Pat DE 2738010, 1978, CA, 1978, v. 88, 169602
-
Pat EP 17171, 1980
-
A. Kurosaki, Chem. Lett.,1988 (1), p 17-20; Pat JP 88264 Du Pont-Mitsui Fluorochemicals
Co Ltd, 1988; CA, 1989, v. 110, 192255
-
A. Henne, I. Shepard , I. Evan., JACS, 1950, 72, p.2577-2579
-
Pat US 2617836, 1952; W.H. Pearlson, L.I. Hals, JACS, 1953, 75, p. 2698
-
R.N. Haszeldine, J. Chem. Soc., 1953, p. 3571
-
Pat SU 150838, 1966
-
Pat. JP 86 277645, 1986; CA, 1986, v.107, 39201
-
Pat. JP 86 221144, 1986; CA, 1986, v.106, 83996
-
Pat US 3213134, 1965
-
Pat US 4302608, 1981
-
Pat EP 54227, 1982,
-
Pat SU 841243, 1996, Vilenchik and others
-
Pat US 3321515, 1967; Pat FR 1416013, 1965
-
Pat JP 78 25512, 1978; CA, 1978, v.89, 59660
-
Pat US 4238416, 1980; CA, 1981, v. 94, 120873
-
Pat SU 740741 ,1980
-
Pat JP 83 62131, 1983; CA 1984, v.100,5862
-
Pat JP 85 188332, 1985; CA, 1986, v.104, 187023
-
Pat SU 859348, 1981
-
Pat JP 83 62130, 1983; CA, 1984, v.100,5861
-
Pat US 3900372, 1975; CA, 1975, v.83, 192571
-
C.Francese, M.Tordeux, C.Wekselman. Tetrahedron Lett.,1988, 29(9),p. 1029-1030;
Pat EP 254652, 1988, CA, 1988, v.109, 210530
-
Pat DE 3816932, 1989; CA, 1989, v.111, 41795
-
Chem. Phys., 1994, 195(8), p.2887-28903; CA, 1994, v.121, 137260
-
Pat US 3367971, 1968; CA, 1968, v.69, 51607
-
I.L Knuniants and others, Izv. Akad. Nauk SSSR, Khimia, 1973(12), p.2725
-
Tetrahedron Letters, 1994, v. 35, 36, p.6721
-
D. Brunel, P. Moreau, J. Burdon, P. Col, J. Chem. Soc, Perkin Trans I, 1989(12),
p.2283-2287; CA, 1990, v.112, 216283
-
Pat US 5629460, 1997; CA, 1997, v.126, 317177
-
J Chem. Soc., 1954, p.1273
-
W.W.Sandler, J. Org. Chem., 1988, 53(1), p. 121-126
-
Pat US 5466879, 1995; CA, 1996, v.124, 145435; PCT Int. Appl. 95 23124, 1995;
CA, 1996, v. 124, 29208
-
A. Elsaesser, W. Sundermeyer, Chem. Ber., 1985, 118(11), p.4553-60
-
Pat US 2614129, 1952; CA, 1953, v. 47, 8769e
|
