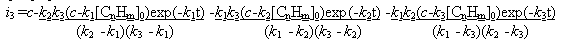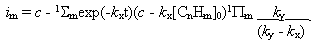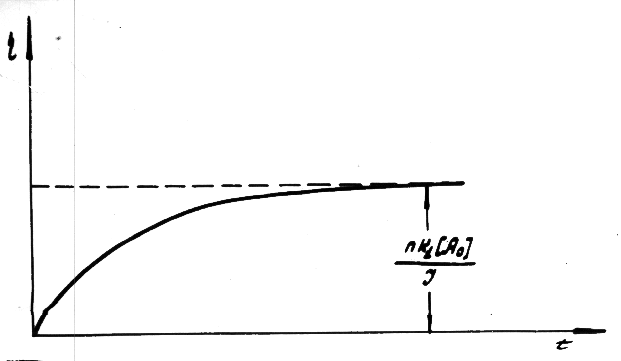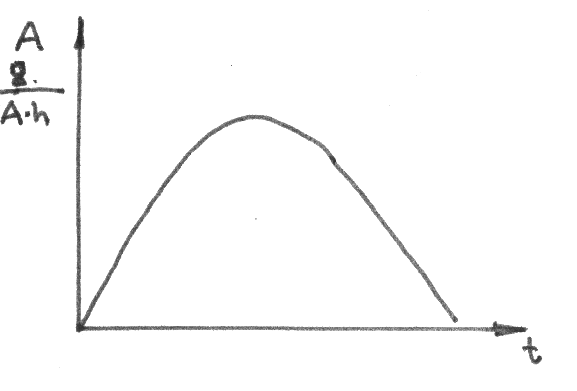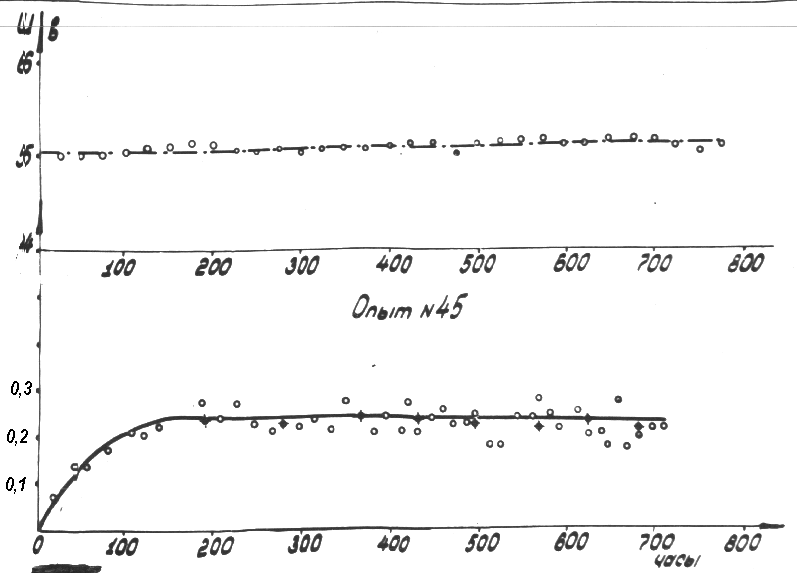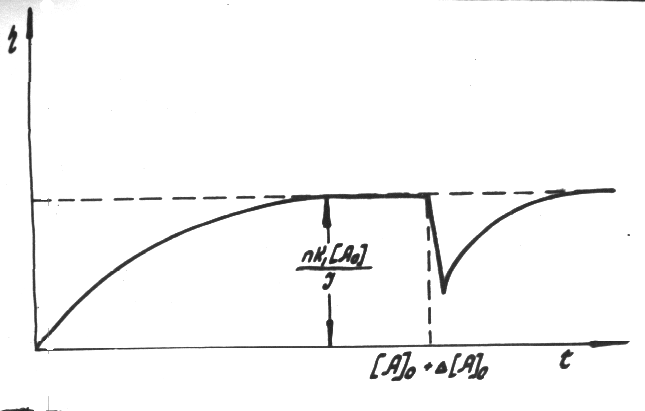
Fig.12 Plot of current yield function at c = k1 [CnHm]0 and [CnHm] = [CnHm]0 +  [CnHm]
[CnHm]
and confirmed by the experimental curve in
Fig.13.
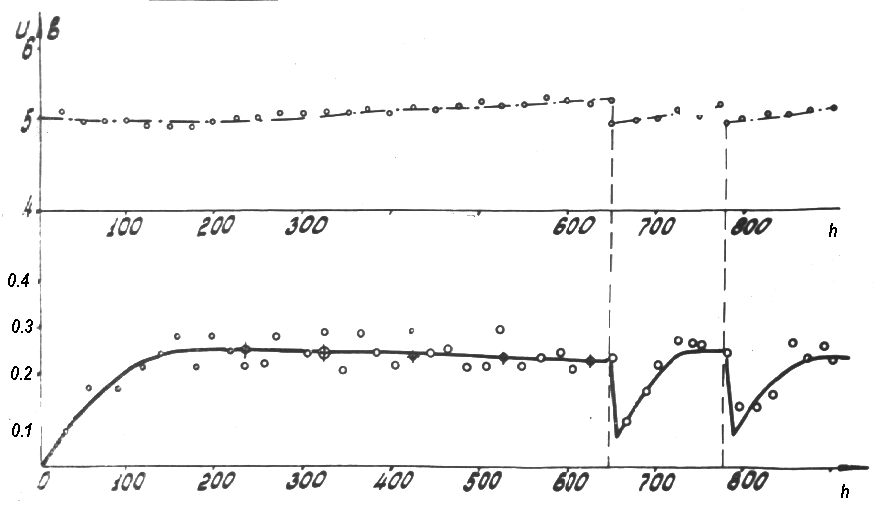
|
Fig.13 Experimental curve of current yield RFCOF (g/A-h) in ECF of CH3OCH2CH2COOCH3 under steady-state conditions after additions of the starting substrate
|
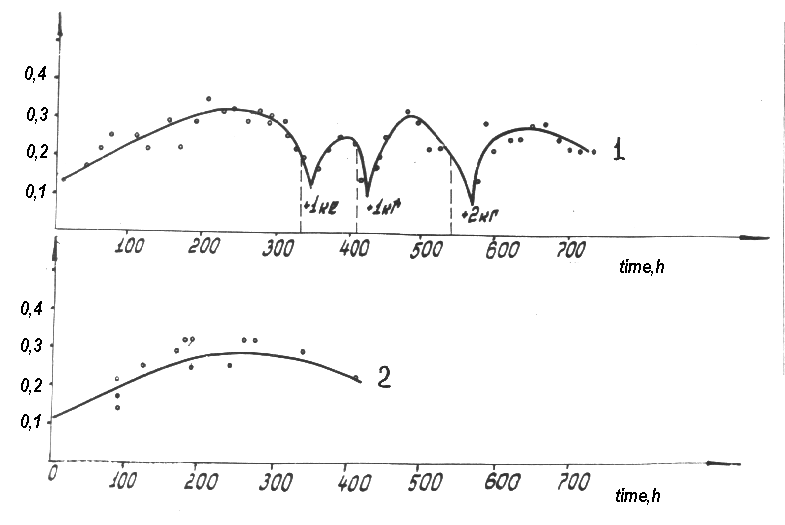
Such an extreme drop in the current yield of the end products at one-time
correction results in a reduction in the average current yield and hence,
in the electrolyzer capacity. Also this brings to a noticeable scatter of
the values to be determined, which is associated with batch nature of the
starting product feed into the electrolyzer in the lasting continuous process.
This phenomena is not observed at continuous feed by a dozing pump.
Overfeeding the starting product resulted also in a reduction in the current
yield due to an accumulation of resin type high molecular products in the
electrolyte, but this is a subject of a special consideration.
Thus, the performed formal kinetic analysis of multistage nature of ECF has been
convincingly confirmed in the electrochemical fluorination of methyl-3-methoxypropionate
and some other oxygen-containing compounds and may be probably used in continuous
electrolysis of different compounds.
Conclusions
-
Usually , dissolution of organic compounds in anhydrous hydrogen fluoride
is accompanied by their protonation with formation of cations which can
not participate the electrode process on the surface of the positively
charged nickel anode.
-
The nickel anodic polarization in AHF medium is accompanied by anode passivation
due to a formation of a nickel fluoride film, on whose surface at high
anodic potentials fluorine ion discharge takes place with adsorption
of fluorine radicals on nickel fluoride that prevents their molization.
-
Evidently, organic compounds in AHF medium are able to be deprotonated forming
complex compounds with nickel fluoride. This determines Simons' ECF specificity
when fluorine radicals in adsorbed state in a phase film of nickel fluoride
interact with the structure of organic molecules.
-
The nature of the organic substrate and its interaction with nickel fluoride
on the anode surface produce a determining influence on processes of
fluorination, destruction and other structure transformations accompanying
ECF of organic compounds.
-
The formal kinetic analysis of the multistage ECF process ,in which slowed
down stages are not only the electrochemical stage of discharge but also
consequent chemical stages of fluorination, indicates that for successful
realization of the lasting continuous process it is required to keep
strictly a steady concentration of the substrate to be fluorinated in
the electrolyte.
References
1. G. Glokler, "Teoreticheskie voprosy v oblasti khimii ftora", "Ftor i ego soedineniya",
pod red. J. Simonsa, IL, Moskva, 1953, 1, 270.
2. V.M. Latimer "Okislitel'nye sostoyaniya ehlementov i ih potentsialy v vodnyh rastvorah",
IL, Moskva, 1954, 396s.
3. U. Sheppard i K. Sharts, "Organicheskaya khimiya ftora", Mir, Moskva, 1972, s.
12-17.
4. M. Stacey, J.C. Tatlow, "Advances in Fluorine Chemistry", London, 1960, v.1, p.
166.
5. F. Lebeau, A. Damiens, Compt. Rend., 1927, 185,652.
6. O. Ruff, W. Mensel, Z. anorg. allg. Chem., 1930, 190, 257.
7. O. Ruff, J. Fischer, F. Luft, Z.anorg.allg.Chem.,1928, 178, 417; O. Ruff, L. Staub,
Z. anorg. allg. Chem., 1931, 198, 32.
8. F. Lebeau, A. Damiens, Compt. Rend., 1926, 182, 1340.
9. J.H. Simons. J. Electrochem. Soc., 1949, 95, N 1, 47.
10. L.P. McCormick, Wisconsin Eng., 1954, N8, 28. 34. 56.
11. I.N. Rozhkov, Usp. Khim.,1976, 45, 1222.
12. H. Schmidt, H.D. Schmidt., Chem. Tech., 1953, 5, 454; J. Prakt. Chem., 1955,
2, 250.
13. W.V. Childs, "Technique of Electroorganic Synthesis", N.L. Weinberg, B.V. Tilak,
eds., John Wiley &Sons, New York, 1982, Vol V, Part III.
14. H. Moissan, Compt. Rend., 1986, 102, 1543.
15. N. Watanabe, Kagaku to koge, Chemistry and Chemical Industry, 1965, 18, N 6,
825.
16. W.S. Fyfe, J. Chem. Phys., 1953, 21 N 1, 2.
17. Hu Jiu-Heng, D. Waite U.Z. Jonston, J.Am.Chem.Soc., 1953. 75, N 1232.
18. W. Klatt, z. anorg. allg. Chem. 1937, 233, 307.
19. I.G. Ryss, "Khimiya ftora i ego neorganicheskih soedinenij", Goskhimizdat, Moskva,
1956. 53.
20. J.H. Simons, J.W. Bouknight, J. Am. Chem. Soc., 1932, 54, 129.
21. J.H. Simons, R.D. Dresdner, J. Am. Chem. Soc., 1944, 66, 1070.
22.К. Fredenhagen, J. Dahmlos, Z. anorg. allg. Chem., 1928, 178, 272.
23. E. Runner,C. Balog, M. Kilpetrick, J. Am. Chem. Soc.,1956, 78, 5183.
24. Kilpetrick,T.J. Lewis, J. Am. Chem. Soc.,1956, 78, 5187.
25. Kilpetrick, "The Structure of Electrolytic Solutions", W.J. Hamer ed., New York,
London, 1959, Part 19, 291.
26. K. Fredenhagen, G. Cadenbach, Z. anorg. allg. Chem., 1928, 178, 272: Z. phys.
Chem., 1930, 146, 245; 1933, 164A, 201.
27. K. Fredenhagen, G. Cadenbach, W. Klatt, Z. phys. Chem.,1933, 164A, 176.
28. A.F. Clifford, H.C. Beachell, W.M. Jack, J. Inorg. Nucl. Chem., 1957, 5, 57.
29. S. Kongricha, A.F. Clifford, J. Inorg. Nucl. Chem., 1961, 18, 270.
30. W. Lange, Z. anorg. allg. Chem., 1933, 215, 321.
31. H.H. Hyman, M. Kilpatrick, J.J. Kats, J. Am. Chem. Soc., 1957, 79. 3668; H.H.
Hyman, R.A. Garber, J. Am. Chem. Soc., 1959, 81, 1847.
32. W. Klatt, Z. anorg. allg. Chem., 1935, 222, 289.
33. W. Klatt, Z. anorg. allg. Chem., 1935, 222, 225.
34. W. Klatt, Z. anorg. allg. Chem., 1937, 232, 393.
35. W .Klatt, Z. phys. Chem.,1935, 173A, 117.
36. W. Klatt, Z. anorg. allg. Chem.,1937, 234, 189.
37. M. Kilpetrick, F. Luborsky, J. Am. Chem. Soc., 1953, 75, 577.
38. K. Wiechert, S. Wies, Univ. Greifswald Math. Natur. Reiche, 1953-1954, 3, 173.
39. J.H. Simons, H.T. Francis, J. Hogg J. Electrochem. Soc., 1949, 95, 53.
40. M.A. Okatov, Zh. Optikomekh. Prom., 1959, N 12, 37.
41. I.L. Knunyants, G.A. Sokol'skij, "Reaktsii i metody issledovaniya organicheskih
soedinenij", Goskhimizdat, 1957. t. 6, str. 343.
42. T. Gramstad, R. Haszeldine, J. Chem. Soc. London, 1956. 173; R. Haszeldine, F.
Nyman, J. Chem. Soc. London, 1956, 2684.
43. S. Nagase, H. Baba, R. Kojima, Repts. Govt. Ind. Res. Inst. Nagoya, 1964, 13,
N1, 20.
44. H. Bode, E. Voss, Z. anorg. Chem., 1956, 286, 136.
45. J. Burdon, J.C. Tatlow, "Advances in Fluorine Chemistry", London, 1960, v.1,
p.129.
46. N.I. Gubkina, S.V. Sokolov, E.I. Krylov, Usp. Khim. 1966, 35, 12, 2219.
47. Ya.N. Voitovich, V.YA. Kazakov, ZhPKh, 1971, 44, 11, 2452.
48. J.H. Simons, R.D. Dresdner, J. Electrochem. Soc., 1949, 95, N 1, 64.
49. A. Engelbrecht, E. Nachbaur, Monath. Chem. (Vien), 1959,
50. H.H. Rogers, S Evans, J.H. Johnson, J. Electrochem. Soc. 1964, 111, N 6, 701.
51. S .Nagase, H. Baba, R. Kojima, J. Chem. Soc. Japan, Ind. Chem. Sec., 1964, 64,
N 12, 2124.
52. K. Fredenhagen, O. Kreft, Z. Elektrochem., 1929, 35, 670.
53. G.G. Koerber, T. De Vries, J. Am. Chem. Soc., 1952, 74, N 20, 5008.
54. J. Sargent, A. Clifford, W. Lemmon, Anal. Chem. 1953, 25, N , 1727.
55. E.A. Ajkazyan, N.M. Arakelyan, S.E. Isabekyan, Izv. AN Arm. SSR, 1964. 17, N
2, 131.
56. N. Hackerman, E.S. Snavely, L.D. Fiel, Electrochem. Acta, 1967, 12, N 5, 535.
57. N. Watanabe, B. Chang, Denki Kagaku, 1969, 37, 198.
58. N. Watanabe, B. Chang, Report on The V Internat. Fluor.Chem. Symp.Moskaw, 1969.
59. G.I. Kaurova, L.M. Grubina, Ts.A. Adzhemyan, Elektorkhim., 1969, 3, N 10, 1222.
60. B. Burrows, R. Jasinsky, J. Electrochem. Soc. 1968, 115 ,N 4, 348.
61. M. Novak and J. Boa, J. Electroanal. Chem. 1980, 109, 179.
62. L.A. Mirkind, Usp. Khim., 1975. 44, N 11, 2088.
63. I.L. Serushkin, G.A. Tedoradze, G.I. Kaurova, G.P. Il'inskaya, T.L. Razmerova,
Elektrohim., 1975, 11, N 5, 705.
64. A.F.Сlifford, A.C. Tolumello, J. Chem. Eng. Data, 1963. 8, N 3, 425.
65. A.F.Сlifford, T.J. Sargent, J. Am. Chem. Soc., 1957, 79, 4041.
66. J .H. Simons, W. Harland, J. Electrochem. Soc., 1949, 95, N 1, 55.
67. S .Nagase, H. Baba, R. Kojima, J. Chem. Soc. Japan, 1961, 64, N 8, 1397.
68. S. Nagase, H. Baba, R. Kojima, J .Chem. Soc. Japan, 1961, 64, N, 2126.
69. S. Nagase, H. Baba, R. Kojima, J. Chem. Soc. Japan, 1962, 65, N 8, 1183.
70. Simons, US Pat. 2.490.098., 6.12.1949.
71. T.C. Simmons, F.W. Hofmann, R.B. Beck, H.V. Holler. T. Katz, R.J. Koshar, E.R.
Larsen, J.E. Mulvaney, К.Е.Раulsen, F.E. Rogers,В. Singleton, R.E. Sparks, J.
Am. Chem. Soc., 1957, 79, N 3429.
72. E.A. Kauck, J.H. Simons, US Pat. 2.644.823. 7.07.1953.
73. V.V. Berenblit, B.A. Byzov, V.I. Grachev, I.M. Dolgopol'skij, Yu.P. Dolnakov,
ZhPKh. 1975, 48, N 3, 709.
74. V.V. Berenblit, V.P. Sass, L.N. Senyushov, Yu.K. Starobin, ZhOrKh, 1976, 12,
N 4, 767.
75. V.V. Berenblit, V.A. Nikitin, V.P. Sass, L.N. Senyushov, Yu.K. Starobin, Yu.V.
Tsyganov, ZhOrKh, 15, N 2, 284.
76. V.V. Berenblit, Yu.P. Dolnakov, G.A. Davydov, V.I. Grachev, S.V. Sokolov, ZhPKh,
48, N 10. 2206.
77. V.V. Berenblit, Yu.P. Dolnakov, G.A. Davydov, S.V. Sokolov, ZhPKh, 1980, 53,
N 4, 858.
78. V.V. Berenblit, Yu.P. Dolnakov, V.P. Sass, L.N. Senyushov, S.V. Sokolov, ZhOrKh,
1974, 10, N 10, 2031.
79. J.A. Young, R.D. Dresdner, J. Am. Chem. Soc., 1958, 80, N 8, 1889.
80. M. Sander, W. Blo chl, Chem. Ingr. Tech. 1965, 37, N 1, 7.
81. V.S. Plashkin, L.N. Pushkina, S.V. Sokolov, ZhOrKh, 10, N 6, 1215.
82. F. Dvorak, V. Dedek, Coll. Czech. Chem. Comm., 1966, 31 N 7, 2727.
83. N. Arakelyan, Arm. Khim. Zhurn., 1971, 24, 679.
84. P. Sartori, W. Habel, J. Fluor. Chem. 1980. 16(3). 265; J. Fluor. Chem. 1981,
18(23). 131.
85. B.B. Damaskin, O.A. Petrij, V.V. Batrakov, "Adsorbtsiya organicheskih soedinenij
na ehlektrodah", "Nauka", Moskva, 1968, s. 246.
86 J.H . Simons, W.H. Pearlson, T.J. Brice, W.A. Wilson, R.D. Dresdner. J. Electrochem.
Soc., 1949, 95. N 1. 59.
87. M.A.Okatov, Him.Nauka i Prom., 1959, 4, 675.
88. N.F. Ryabinin, I.P. Kolenko, B.N. Lundin, I.V.Bunina, Trudy Inst. Khim. UFAN
SSSR, 1968, N 15. 67
89. R. Kojima, T. Hayashi, S. Takagi, Nagoja Koge Giduzu Sikense Hakoky, 1961, 9.
N 10. 16.
90. H.Kisaki, S.Mabuchi, T.Sakamura, Denki Kagaku, 1966,34.N1.24.
91. H.M.Scholberg, H.G.Bryce, US Pat., 2.717.871, 13.09.1955.
92. V.V. Berenblit, B.A. Byzov, I.M. Dolgopol'skij, Yu.P. Dolnakov, ZhPKh, 1974,
47, N 11, 2433.
93. T. Gramstad and R.N. Haszeldine J. Chem. Soc. 1956. 1, 173;1957. 2640.
94. T.J. Brice and P.W. Trott US Pat. 2.732.398 24.01.1956
95. J. Burdon, J. Farazmand, M. Stacey and J.C. Tatlow, J. Chem. Soc. 1957. 2574
96. T. Gramstad, Tidsk.Kjemi, Berg. Met., 1958, 18, N 10, 157.
97. A.R. Diesslin, E.A. Kauck, J.H. Simons, US Pat. 2.593.737.22.04.1952.
98. G.P. Gambaretto, G. Troilo, M. Napoli, La Chim. L'Ind., 1970, 52, N 111097.
99. I.M. Dolgopol'skij, A.V. Tumanova, V.V. Berenblit, B.A. Byzov, Avt. Svid. SSSR
N 405.326b 01.07.1960.
100. S. Nagase, R. Kojama, Bull. Chem. Soc. Japan, 1961, 34, N 10, 1460.
101. S. Nagase, H. Baba, R. Kojima, Bull. Chem. Soc. Japan, 1962, 35, N, 1907.
102. J.H.Simons, US Pat. 2.500.388. 14.03.1950.
103. S.I. Gerasimov, S.A. Mazalov, V.S. Plashkin, S.V. Sokolov, ZHOH, 1966, 36, N
3, 532.
104. V.S. Plashkin, L.N. Pushkina, S.L. Mertsalov, V.F. Kollegov, S.V. Sokolov, ZhOrKh,
1970, 6, N 5, 1006.
105. V.S. Plashkin, L.N. Pushkina, Yu.P. Dolnakov, S.V. Sokolov, ZhOrKh, 1973 ,9,
N 12, 2525.
106. S.A. Mazalov, S.T. Gerasimov, S.I. Sokolov, V.A. Zolotarin, ZhOKh, 1965, 35,
N 3, 485.
107. H. Meinert, R. Fackler, J. Mader. P. Reuter, J. Fluor. Chem., 1991, 51, N1,
53; 1992, 59, N,351.
108. V.Ya. Kazakov L.A. Savel'ev, R.A. Dzerzhinskaya, U.A. Shishkin, I.L. Gudilov,
ZhPKh, 1968, 41, N 102212, 2220.
109. A. Dimitrov, W. Radeck, Sruediger, O.Berstein, J. Fluor. Chem., 1993, 60, N
1, 57.
110.А.F. Clifford, H.K. El-Shamy, H.J. Emeleus, R.N. Haszeldine, J. Chem. Soc. London,
1953, 2372.
111. T.C. Simmons, F.W. Hofmann, R.B. Beck, H.V. Holler. T. Katz, R.J. Koshar, E.R.
Larsen, J.E. Mulvaney, К.Е.Раulsen, F.E. Rogers,В. Singleton, R.E. Sparks, J.
Am. Chem. Soc.,1957, 79, N ,3426.
112. W.H. Pearlson US Pat 3.274.081, 20.05.1966, Brit. Pat 1.007.288., 1965
113. S. Nagase, H. Baba, R. Kojima, Bull. Chem. Soc. Japan, 1962, 35, N 1, 29.
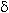 [CnF
îm-1]/
[CnF
îm-1]/