Synthesis and application of 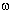 -bromoperfluoroalkylvinyl
ethers -bromoperfluoroalkylvinyl
ethers
V.M. Andrushin, N.B. Pavlova
Report 1. New method to synthesize -bromo-perfluoroalkylvinyl
ethers.
Introduction.
In recent decades fluorine containing plastics and rubbers have found wide application
in different fields of technique requiring high performance of composite
materials(thermal and chemical stability, resistance to acids, industrial
liquids, solvents, alkalis). The necessary physicochemical, physicomechanical
and other characteristics are provided by creation of copolymers of various
types.
Conventional homo- and copolymers on base of fluorine-containing olefins
such as tetrafluoroethylene (TFE), vinylidene fluoride (VDF), perfluoroalkyl
ethers have a number of known disadvantages. That is why fluoromonomers
on the same basis containing different functional groups have recently
attracted an increased interest. They possess positive properties of
fluoroplasts excelling them in a number of such characteristics as hydrophily,
electroconductivity, solubility, processibility, anti-adhesion and others.
The analysis of data from scientific -technical and patent literature has
shown that a great part of modifiers consists of fluorinated vinyl ethers
of the following general formula :
CF2 = CFORfX
where Rf is fluoroalkyl fragment, X is different functional substituents(Cl
, Br , CN , Y , C(O)OR, OC6 H5, C(O)CF3,
SO2F , OCF=CF2, CF-CF2 and others).
The content of perfluoroalkylvinyl ethers in such polymers is relatively small
but it makes the properties of copolymers significantly vary.
Sometimes to solve specific problems, a third monomer with similar properties
is introduced to make individual types of polymer materials varying such
additives as cross-link agents, fillers, plasticizers etc.
-
bromo-perfluoroalkylvinyl ethers (BrAVE) are convenient base reagents to
create new classes of fluoropolymers with different functional groups,
as well as modifiers for fluoropolymers.
It has been determined experimentally that BrAVE are easy to copolymerize
with TFE, VDF and other fluoroolefins by conventional methods. In this
case the content of bromo-monomer in the copolymer amounts to 26%.
Modification of fluoroplasts may be accomplished by introducing BrAVE into
reaction of copolymerization with TFE and other fluoroolefins followed
by its cross-linking to make cross-linked fluoropolymers. Another modification
method is using perfluorodivinyl ethers with different chain length in
copolymerization. These ethers are easy to synthesize from appropriate
BrAVEs.
To create special fluoropolymers with active centers, for example for ion-exchange
membranes (IEM), it is difficult to attain desired results by copolymerization
of fluoroolefins with fluorinated vinyl ethers containing the appropriate
functional groups because of low reactivity of the latter. Therefore
the copolymerization of fluoroolefins with BrAVE followed by bromine
replacement with specific functional groups by means of polymer-analogous
conversions seems perspective. Effectiveness of this method has been
confirmed experimentally during processing granules and films for IEM
synthesis with anion-active (alkyl-amine) and cation-active (carboxyl)
groups.
In the report we briefly describe an original method developed by the authors
to make BrAVE with different chain length.
In further reports we are going to give main routes for application of BrAVE
monomers and some properties of fluoropolymers modified by them and possible
fields of their application.
Principal stages of the process.
BrAVEs have been described in patent literature (Japan) as crosslinking monomers
as a compound of the general formula of Br(CF2)nOCF=CF2 , while synthesis
methods and properties of every specific compound have not been given.
We have selected BrAVEs with n=2 and n=4 as the most perspective and developed
a fundamentally new synthesis method according to the following scheme:
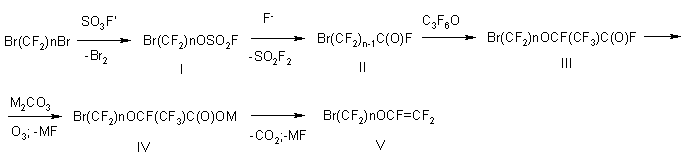
Main raw material in the synthesis of BrAVE are
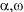 -
dibromoalkyles with different chain length ( a method of their production
is going to be described in further reports), covalent fluorosulfonates
and hexafluoropropylene oxide. -
dibromoalkyles with different chain length ( a method of their production
is going to be described in further reports), covalent fluorosulfonates
and hexafluoropropylene oxide.
The synthesis of BrAVE monomers in this report is described taking as example
R-114b2 (1,2-dibromtetrafluoroethane), the most available refrigerant
in Russia, and R-318b2 ( dibromodecafluorobutane).
Synthesis of
-bromo-perfluoroalkylfluorosulfates.
To produce monofluorosulfonates (1), covalent fluorosulfonate (peroxydisulfuryldifluoride,
S2O6F2; chlorine fluorosulfonate, ClOSO2F;
bromine fluorosulfonate, BrOSO2F or their mixtures) is added
slowly to the starting dibromoalkane ( R-114B2 or R-318B2 respectively)
at vigorous stirring at a temperature of 40-50
o
C at a mole ratio of 1:1-3:1.
To initiate the process, a catalytic amount of bromine is added for more
active formation of bromine fluorosulfonate:
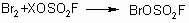
,
X= Cl- , -OSO2F.
To optimize the process, the order of mixing and ratio of reagents have been
investigated. According to our data, an addition of dibromoalkane to the
fluorosulfatating mixture is inexpedient because of great exothermicity of
the reaction and difficulties in heat removal.
The optimal mole ratio of FSC-PSF/dibromoalkane is 1.3:1. Further increase
in amount of fluorosulfatating agent leads to an increase in the content
of di-substitution product (bis(fluorosulfonyloxy)perfluoroalkane)-BIFS
in the reaction products:
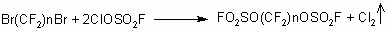
A prolonged boiling of the reaction mixture gives a similar result.
The content of the reaction products versus the ratio of the reagents is
plotted in Fig.1
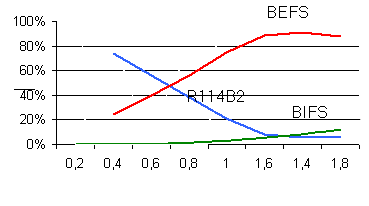
Fig.1 Yield of mono-and di-substituted products of fluorosulfatation of R-114B2
versus the ratio of the reagents.
The content of the goal product in the reaction mixture under optimal conditions
was 92-95%.
Elemental bromine and chlorine are formed together with the goal product
as a result of the reaction. Chlorine is blown away during the synthesis
to an absorbing column, bromine and unreacted fluorosulfonate can be
removed from the reaction mass by means of a solution of soda or alkali,
but it is more reasonable to treat the reaction mass with tetrafluoroethylene
at stirring and cooling. At that in case of bromoperfluoroethylfluorosulfate,
the starting R-114B2 is recovered and additionally the goal product is
formed:
Br2
+ CF2
=CF2
-> BrCF2
CF2
Br
BrOSO
2
F + CF2
=CF2
-> BrCF2
CF2
OSO2
F
The reaction products were separated on a packing rectification column, the
yield of the goal products was up to 90%, and the purity was up to 99-99,5%.
Synthesis of fluoroanhydrides
of -bromoperfluorocarbonic acids.
Fluoroanhydrides of
-
bromoperfluorocarbonic acids(2) were prepared by decomposition of alkylfluorosufates
in a medium of a polar aprotic solvent (diglyme, adipodinitrile) in the
presence of anhydrous fluoride of alkali metal (K,Na,Cs).
Decomposition of
-
bromoperfluoroethylfluorosulfate (BEFS) was carried out at room temperature
at a mole ratio of KF:BEFS=0.15-0.5:1 in a flask equipped with a stirrer
and a cooler cooled to minus 50
o
C. At that the forming fluoroanhydride of
-
bromoperfluoroacetic acid (b.p.=0- -1
o
C) was recycled to the reactor and sulfuryldifluoride ( by-product) was condensed
in a trap (-78
o
C).
After the completion of decomposition, the reaction mass was heated to 50
o
C without cooling the cooler and the producing fluoroanhydride of
-
bromoperfluoroacetic acid was condensed in a trap (-78
o
C) and then transferred into a cylinder.
The investigations made have shown that such a procedure to eliminate fluoroanhydride
of
-
bromoperfluoroacetic acid makes possible to attain the maximum yield of the
product (90%). Another method, producing and storage of producing fluoroanhydride
of
w
-
bromoperfluoroacetic acid as potassium alkoxide in a solution of diglyme
or ADN, leads to significant losses due to alkoxide dissociation and
low boiling point of the fluoroanhydride.
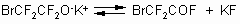
Synthesis of fluoroanhydride of
-bromoperfluorobutyric acid was carried out similarly at a temperature of
decomposition of 50-60oC. The goal fluoroanhydride was distilled
from the reaction mass ( b.p.=57-58oC), purified by rectification.
The yield was 85-90%.
Synthesis of fluoroanhydrides of -bromo-2-trifluoromethyl-3-oxaperfluorocarbonic
acids.
The synthesis of compounds (3) was carried out according to a classical scheme
widely used in laboratory practice by means of hexafluoropropylene addition
to fluoroanhydrides of appropriate perfluorocarbonic acids(2). The reaction
is carried out in the presence of anhydrous fluorides of alkali metals
in a medium of different polar aprotic solvents: glymes, nitriles, tetrahydrofurane,
dimethylsulfoxide etc.. Diglyme and tetraglyme possessing higher solvating
ability are used most often.
However the conducted experiments have shown that the use of diglyme leads
to a partial substitution of the bromine atom in the molecule of addition
product (3) and to the formation of fluoroanhydrides of perfluoropolyoxa-carbonic
acids in the reaction products. Analysis of the solid residue of the
reaction mass has shown the presence of bromine-anion.
Therefore as a solvent in the stage of HFPO addition we used adiponitrile
dried by distillation under vacuum over P2O5.
The reaction was carried out in two ways:
1.Alkoxide was produced at atmospheric pressure, stirring, room temperature
then HFPO was bubbled into the reaction mixture at 0-5oC:
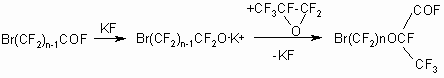
This method is laborious, requires a long time and is distinguished
by low productivity and not high enough yield, especially
in case of low-boiling products (n=2).
2
.A method of HFPO addition under pressure is more effective. KF and ADN were
charged into a reactor, evacuated, the starting fluoroanhydride was added,
the reaction mixture was stirred for 2-3 hours to produce alkoxide, cooled
with liquid nitrogen and HFPO was added. The reactor was gradually heated
to room temperature at stirring to the pressure drop stopping.
The optimal mole ratios were: KF:FA=0.5-0.8:1; HFPO:FA=1.2:1
After the synthesis completion the contents of the reactor was poured out
to a separating funnel, the lower fluoroanhydride layer was separated
and distilled to fractions. The content of the goal addition product
in the mixture was 60-70 vol.%. The admixtures were HFPO dimer and a
product of addition of two molecules of HFPO. After rectification the
goal product was separated with 98-99% purity.
Production of fluoroanhydrides of
-bromoperfluorocarbonic acids (2) and products of HFPO addition to them (3)
is carried out under similar conditions in a medium of dehydrated aprotic
solvent in the presence of fluoride of alkali metal. Therefore there
was investigated a possibility (and expediency) to carry out both stages
in the same reactor unit in different variants described by the general
scheme:
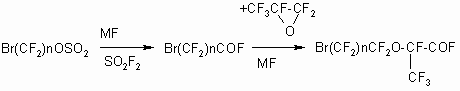
There are two ways to realize the scheme:
The first one: to separate intermediate fluoroanhydride of w-bromoperfluoro-carbonis
acid with following addition of HFPO to it.
The second way is HFPO feeding into the reaction mass produced in the previous
stage without fluoroanhydride (3) separation with further separation
of the goal product.
Experiments have shown that the scheme of consecutive synthesis is quite
acceptable to produce small laboratory samples, but in this case a decrease
in the yield of the goal product is inevitable because one of the reactions
is carried out under nonoptimal conditions. However here equipment get-up
is easier and the use of low-temperature reagents to separate the intermediate
anhydride is excluded.
To produce a very pure product of HFPO addition in big quantities, it is
reasonable to use the first scheme with intermediate separation and using
optimal reaction media. But here a special equipment is necessary to
provide low-temperature collection of
-anhydride(3) and then HFPO addition at an elevated pressure.
Production
and purification of salts of -bromo-2-trifluoromethyl-3-oxaperfluorocarbonic acids.
The goal
-bromoperfluoroalkylvinyl ethers were produced by pyrolysis of addition products
(4) in different ways: pyrolysis of a dry salt produced preliminarily or
liquid pyrolysis of a salt produced directly in a solvent.
Dry salts (4) were produced by hydrolysis and neutralization of addition
products (3) with aqueous solution of KOH,NaOH,Na2CO3.
The solution was evaporated to dryness, the residue was pounded with
pestle, water traces were removed by azeotrope distillation with
benzene, the dry salt after repeated pounding with pestle was dried
under vacuum (5-10mm mercury column) at a temperature of 110-120oC
for 5-10 hours.
The absence of moisture is the necessary condition of the pyrolysis.
Otherwise the formation of side hydrogen-containing products, hydrides,
occurs.
Method to produce and purify
salts of -bromo-perfluoro-alkyloxaalkylen carbonic acids on a laboratory
scale.
Sodium salt of
-bromo-perfluoro-2-methyl-3-oxavaleric acid was taken as an example.
It was synthesized from the appropriate fluoroanhydride by its treatment
with soda in the presence of catalytic amounts of water.
It is easy to produce the mentioned salts in principle by consecutive
hydrolysis of the appropriate fluoroanhydrides and treatment of the
acid produced with soda. But it is rather difficult to purify the
salt produced from hydrogen fluoride and residue moisture. Hydrides
retard the copolymerization process of fluorovinyl ethers with fluoroolefins,
therefore the content of hydrides in the goal product is strictly
limited: 0.1%. Moreover, hydrides form azeotropes with vinyl ethers
that makes difficulties for their purification and reduces the yield.
Some ways of technological routes have been studied:
a). Interaction of the fluoroanhydride with crystal hydrates of the following
composition: Na2CO3*5H2O. The salt
formation runs very smoothly; to remove moisture, salt was first
dried by common methods and the crushed salt was then evaporated
at heating to 100-120oC.
b). Mixing fluoroanhydride, soda and catalytic amounts of water in different
combinations with use of benzene. Here moisture is easy to remove
as azeotrope with benzene.
Both methods can be used to make the salts of necessary quality. Using
different methods of purification, samples of sodium, potassium,
lithium salts of 5-bromo-perfluoro-2-methyl-3-oxavaleric acids were
produced and tested during the pyrolysis process.
It has been found during the producing of aggregative samples, that hygroscopicity
of K-salts does not allow to dry them by the mentioned methods to
a sufficient degree.
Producing -bromo-perfluoroalkylvinyl
ethers.
I
nfluence of the composition and quality of the salts on the quality and
yield of fluoroalkylvinyl ethers has been determined during this
study.
Dry samples of sodium, potassium and lithium salts were subjected to
pyrolysis at a temperature up to 300oC. At a temperature
up to 120-140oC there was no evidence of decomposition
of the salts. At 160oC the pyrolysis degree does not exceed
5% that is due to some local overheating in our opinion. At a temperature
above 300oC sometimes uncontrolled reaction took place
accompanied with self-heating the reaction mass (made red-hot) and
complete destruction of the reagents. Therefore the yields of the
goal monomer were determined at two temperatures: 180oC
and 250oC.
The yield of the monomer was calculated according to its content in the
pyrolyzate condensed in traps cooled to -78oC.
The pyrolyzate was up to 96-97% of the goal product under the optimal
conditions. Further purification was carried out by rectification
on a "Perkin-Elmer" preparative column. The yield of vinyl ethers
was 87-90% and the purity of 99.5-99.9%.
The yield of the pyrolyzate at a temperature of 180-250oC
differs by 0.8-1.8% that is within the experimental error, that means
that the yield does not practically depend on the temperature.
In a row Li-Na-K the yield of the goal monomer is increasing from 66-68%
to 85.0-85.5% that is in accordance with the size of the cation and
the data on hygroscopicity of the salts.
The liquid pyrolysis makes it possible to reduce the reaction temperature
to 140-180oC. In this case requirements for drying the
starting components remain the same. A solvent, diglyme or adiponitrile,
was preliminarily kept over drying agent under vacuum. Soda or potash
was calcined at 250-300oC in vacuum with periodical pounding.
The method consists in a slow addition of fluoroanhydride to a suspension
of carbonate in a solvent at vigorous stirring and cooling to 10-15oC.
The control over the salt formation is executed by intensity of carbon
dioxide liberation. After the completion of salt formation the temperature
is slowly raised to 120-140oC and then to 180oC
condensing the pyrolysis products in traps (-78oC). The
content of the goal product in the pyrolyzate is 80-90% and the yield
is 85-90%.
The individuality of the compounds produced has been determined by GLC
method, the structure has been confirmed by 19FNMR, 1H proton NMR,
IRS analyses.
A choice of the solvent (diglyme, acetonitrile,adiponitrile) in the pyrolysis
process is of great importance.
Diglyme and adiponitrile have been found to be the most suitable and
the process runs much actively in the first one, but it is necessary
to take into account complex influence of salt-solvent couple to
make the final choice of the pyrolysis conditions.
The produced samples of
-bromoperfluoroethyl- and
-bromoperfluoro-butylvinyl ethers with a purity of 99.5-99.8% were
used as co-monomers with tetrafluoroethylene and vinylidene fluoride
to modify fluoroplasts and synthesize fluoropolymers with functional
groups.
Conclusions.
The framework of the paper does not allow to describe in detail all features
of all the stages of the process of BrAVE synthesis, but the authors
believe that the above information is quite enough to evaluate effectiveness
and perspective of the proposed synthesis method for monomers of
new class which can solve the problems of fluoroplasts modification
and creation of new perspective fluoropolymers with different functional
groups.
Specific methods and ways to synthesize semi-products should be chosen,
traditions and specific opportunities of research organizations in
raw materials and equipment taken into account. Moreover, one should
have in mind that both the goal
-bromo-perfluoalkylvinyl ethers and bromine-containing intermediate compounds
are high-toxic substances. They belong to the 2nd class
of danger and require the appropriate safety and protective measures.
|
