Interactions of asymmetric perfluorosubstituted 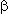 -diketones and carbamoyl amines. -diketones and carbamoyl amines.
L.M. Popova, A.Yu. Trishina
Saint-Petersburg State Technological Institute (Technical University)
26, Moskovsky ave., Saint-Petersburg, 198013, Russia
department of molecular biotechnology
A cyclocondensation reaction of asymmetric perfluoro-substituted
-diketones with derivatives of guanidine and urea has been investigated. There
have been determined conditions of the formation of functional derivatives
of 6-perfluoro-substituted 1,3-diazines
.
Organofluoric compounds are used for producing thermally and chemically stable
polymers, medicines, pesticides, high-resistant dyes, refrigerants and heat
carriers, lubricants and other important products. The creation of heterocycles
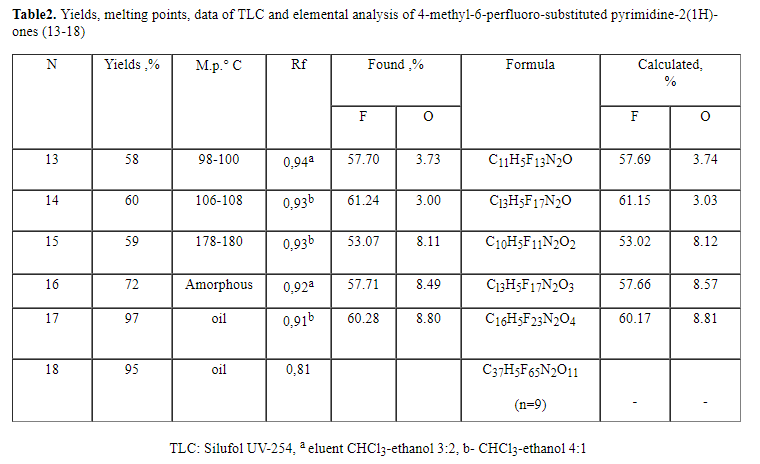
whose side chain containins inert perfluorosubstituents with a large number
of units is of a practical interest. This is caused by a well-known fact,
that the series of perfluorocontaining carbon-chain and heterochain compounds
( for example, perfluorocarbonic acids) possess pronounced surface-active
properties enhanced with the growth of the alkyl chain length .
One of the widely used synthesis methods for pyrimidine derivatives is condensation
of 1,3-diketones ( or their analogues) with different derivatives of guanidine
and urea. The range of compounds produced in such a way is extremely wide
[1-3, 4-11]. Thus, condensation of guanidinium hydrochloride with acetylacetone
at heating in a water-alcohol solution of sodium hydrocarbonate results in
the formation of 2-amino-4-dimethylpyrimidine; 2-amino-4,5,6-trimethylpyrimidine
is formed in sodium ethylate; 2-amino-4,6,-dimethyl-5-phenylazopyrimidine
is formed in an alcohol solution of sodium hydroxide guanidine nitrate with
3-phenylhydroazonoacetylacetone [3-5]. T.Nishivaki et al. [4,5] noted that
melting guanidine carbonate with derivatives of 3,3,3-trifluoroacetylacetone
[4] and its nonfluorinated analogue [5]at 140-150oC for 1 hour
results in the formation of appropriate 4-subsituted 2-aminopyrimidines (79-90%).
Another example [6] of synthesis of 6-trifluoromethyl derivatives and 6-heptafluoropropyl
derivative of 4-alkylpyrimidine (Alk = C1 -C5) may be ethylguanidine cyclization
with corresponding 1,3-ketones in a mixture of ether and alcohol at room
temperature for 14 hours (24.7-62.8%). Kreutzerberg et al. [10] also conducted
condensation of 4-tolylguanidine with fluorine containing
-diketones in the presence of sodium carbonate (90-115oC, 8-12 hours)
resulting in derivatives of 6-alkyl-4-trifluoromethyl-2-(4-toluidino)pyrimidine
(Alk =C1-C5), yields reached 49-69%. In 1993 the same authors [11] synthesized
a number of derivatives of 2-[N-(2-hydroxyethyl)methylamino]-4-trifluoromethylpyrimidine
possessing physiological activity by a reaction of sulfate of N-(2-hydroxyethyl)-N-methylguanidine
with various
-diketones with trifluoromethyl subsituent in a water-alcohol solution of sodium
bicarbonate.
Sareen et al. [12] reported on the synthesis of fluoro-substituted pyrimidines,
in particularly on the condensation of asymmetric perfluoro-substituted 1,3-diketones
(RF=CF3, C2F5,C3F7) with guanidinium carbonate at boiling in absolute
alcohol with addition of catalytic amounts of hydrochloric acid during 10-14
hours, as a result 6-alkyl(perfluoroalkyl)-4-arylpyrimidines are formed in
high yields (73-82%). Interaction of the mentioned 1,3-diketones with pentafluorophenylhydrazine
under the same conditions leads to the formation of appropriate 5-perfluoroalkyl-3fluoroaryl-1-pentafluoroarylpyrazoles
(70-80%).
There are data [13] on the synthesis method to produce mono-and ditrifluoromethyl-
and p-fluoroaryl-substituted 2-aminopyrimidines by condensation
of aminoguanidine derivatives with different fluorine containing
-diketones in the presence of sodium in an alcohol solution at heating for 12
hours (54-82%).
Interaction of urea, its methyl-N,N'-dimethyl derivative and also thiocarbamide
with pentane-2,4-dione and its trifluoromethyl analogues in the presence
of hydrochloric acid results in the formation of appropriate derivatives
of 2-hydroxy- and 2-mercaptopyrimidines [14]. Condensation of benzylthiocarbamide
with a number of asymmetric
-diketones containing trifluoromethyl and heptafluoropropyl groups at heating
(aqueous K2CO3, ethanol, ether, 96 hours) led to the formation of appropriate
perfluoroalkyl-2-benzylthiopyrimidines (31-53%) with the exception of 2-benzylthio-4,6-di(trifluoromethyl)pyrimidine
whose yield was 0.3% [7].
Koukhar' et al. [15] have shown that
-alkoxyvinyltrifluoromethylketones are suitable reagents for the synthesis of
4-trifluoromethylpyrimidines unsubstituted in 5 and 6 positions. The interaction
of
-ethoxyvinyltrifluoromethylketone with ammonium chloride in formamide (160oC,
2 hours) leads to the formation of 4-trifluoromethyl-pyrimidine (23%). A
treatment of organofluoric substrate with gemdiamino compounds allows
to obtain 4-trifluoromethylpyrimidines containing various functional groups
(OH,SH,NH2) in position 2. 2-hydroxypyrimidine is formed at heating butenon
with carbamide (120-130oC). Its yield is increasing from 35 to
75% in the course of the reaction during 2 days at 205oC, similarly
2-mercaptopyrimidine (65%) is formed in the reaction with thiocarbamide (H+,
20oC, 2 days). 2-amino-pyrimidines are formed by condensation
of butenon with guanidine and its sulfoderivative (NSO2C6H4NHCOCH3-4) . In
the reaction with guanidinium chloride (EtOH,NaOEt, 60oC, 2 hours),
pyrimidine yields in 45% and heating with guanidinium carbonate in benzene
(120oC, 6 hours) increases the yield to 60%.
The aim of the study was to develop technologically acceptable synthesis ways
and study the physicochemical properties of the new group of functional derivatives
of 6-perfluoro-substituted pyrimidines.
Given the interaction of asymmetric perfluoro-substituted
-diketone(1) with thiocarbamide under conditions of acid catalysis as an example,
kinetic evaluation of the cyclocondensation reaction was performed by spectrophotometric
method by means of registration of the running values of the optic density
index of solutions (D). To measure the optic density of 1,1,1,3,3-pentahydroperfluorodecane-2,4-dione
(1) there were used water-alcohol (isopropanol) solutions of hydrochloric
acid due to low solubility of the diketone (1) in water. The condensation
reaction was carried out at fixed values of temperature of 20and 30oC
in a 50% vol. of thiocarbamide ( at a ratio of the reagents diketone/thiocarbamide
of 1:50) in a 0.1 M solution of HCl (pH 3). The experimental results are
presented in Fig.1.
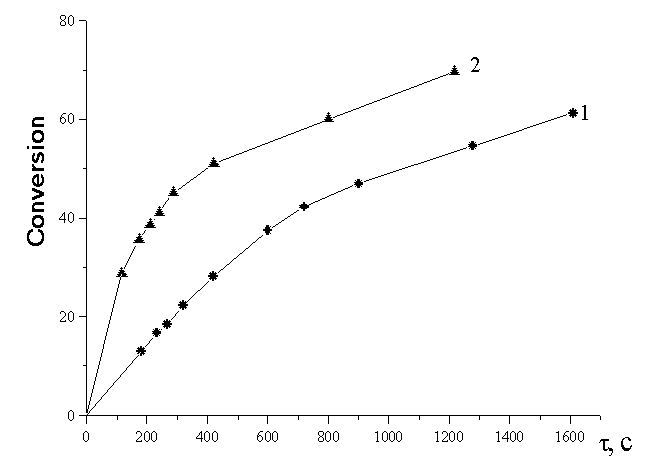
Fig.1. Kinetic curves of the acid cyclization of 1,1,1,3,3-pentahydroperfluorodecan-2,4-dione
(1) (c 10-4 mol/l) and thiocarbamide
The reaction of cyclocondensation is known to proceed in two stages with subsequent
removal of two water molecules [14]. A strong electron-deficient effect of
the perfluoroalkyl substituent causes prevalence of the enol form of the
diketone ( up to 100%) [16,17]. In the first stage, thiocarbamide is added
to the enolizated carbon atom of
-diketone. The removing water molecule is [18] a good leaving group. Further
as a result of intramolecular condensation, a cyclic product is formed (20).
Under these conditions in all the cases considered the dependence of the concentration
of 2-mercapto-4-methyl-6-perfluoropyrimidine (20) on time of cyclization
was satisfactorily approximated with the first order kinetic equation. The
calculation of the cyclization rate was performed according to the first
order equation [19]:
Here 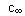 is the concentration of pyrimidine
at reaction completion, ci is the concentration of pyrimidine
at instant is the concentration of pyrimidine
at reaction completion, ci is the concentration of pyrimidine
at instant 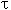 . .
The least-square procedure was used to calculate the values of the pseudomonomolecular
(apparent) rate constants of the cyclization reaction , their logarithms
and half-lives as follows: at 298K
k =0.745
*
103
/font>-1 (ln k 6.613),
1/2 930 c
, at 303K k=0.2.39*103 c-1 (ln k 7.779),
1/2 -300
.
The kinetic evaluation of the reactivity of
-diketone (1) allows to determine the conditions of cyclization with cabomoyl
amines.
As a result of the study, a large number of functional derivatives of pyrimidine
was synthesized by the reaction of cyclocondensation of asymmetric perfluoro-substituted
-diketones (1-6) with guanidinium carbonate, carbamide and thiocarbamide under
conditions of acid catalysis. As a result, the appropriate 6-perfluoro-substituted
2-amino- (7-12), 2-hydroxy- (13-18) and 2-mercapto-(19-24)-4-methylpyrimidines
were found to be formed:
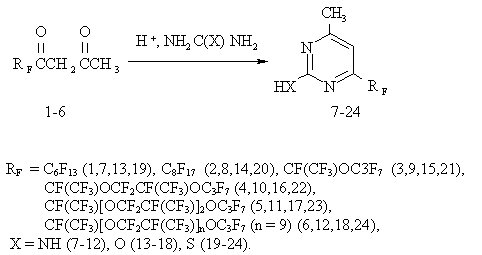
Asymmetric perfluoro-substituted
-diketones (1-6) were used as a dicarbonyl fragment.
The reaction was carried out in a polar solvent ( alcohol) in the equimolar ratio
of
-diketone (1-6) and carbomoyl amine in the presence of an acid catalyst at heating
(70-100oC) for 3-5 h. The yields of 6-perfluoro-substituted 4-methylpyrimidines
(7-24) attained 68-95%. An increase in the yield is connected with partial
solubility of some 6-perfluoro-substituted pyrimidines in a water-alcohol
solution.
Also, 2-amino-, 2-hydroxy- and 2-mercapto-4-methyl-6-perfluorohexylpyrimidine
(7,13,19) were produced in isopropanol at room temperature after 3 days,
the yields of the polycondensation products (7,13,19) were 68, 72 and 72%
respectively. The course of the reaction was controlled by TLC procedure
according to the starting diketone disappearance. Under the conditions of
cyclocondensation of
-diketones with guanidinium carbonate, carbamide and thiocarbamide, attempts
to record chromatographically the formation of the reaction products by only
one ketonic group failed.
Separation of the synthesized compounds was carried out after addition of alkali
or soda solutions (13-24) by distillation of the solvent with subsequent
reprecipitation from water (7,8), vacuum sublimation (13,14), extraction
by ether (19-24), recrystallization from alcohol (9-12, 15-18, 21-24) or
from acetone (7,8,19,20). Perfluoro-substituted functional derivatives of
pyrimidine are separated as crystals (7,9,13,15,19,21), amorphous (10,11,16,20,22)
and viscous oily substances (8,12,17,18,23,24) (Tables 1-3). An attempt to
crystallize compound (20) via fractional vacuum distillation resulted in
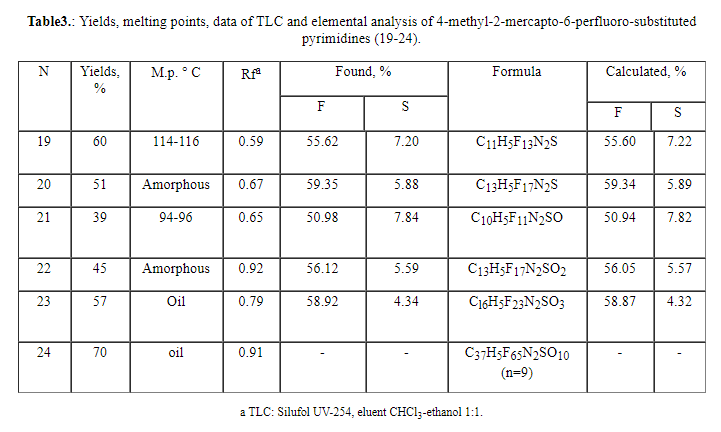
the product decomposition. All synthesized 2-mercaptopyrimidines (19-24)
possess a specific odor.
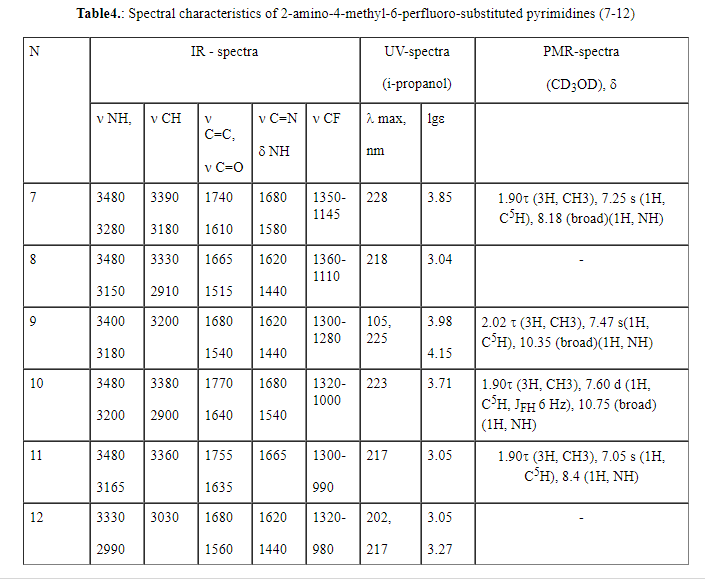
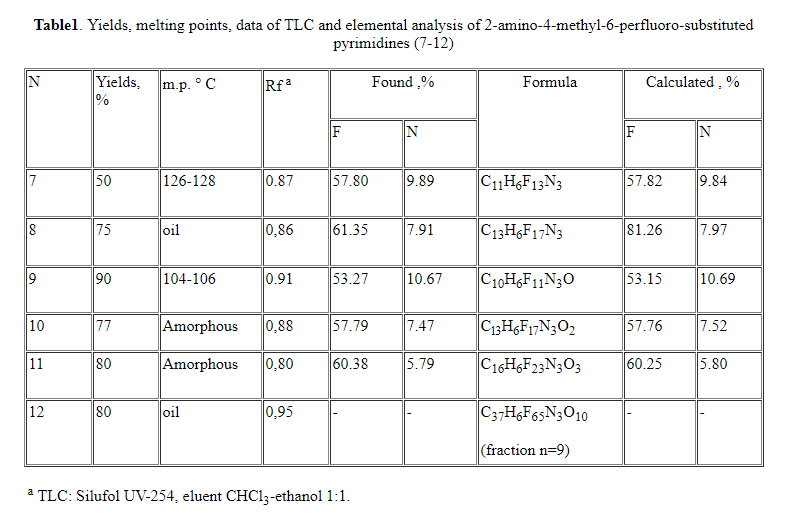
The composition and structure of compounds (7-12) have been confirmed by the
data of elemental analysis, and also by IR, UV and PMR spectroscopy (Tables
4-6).
IR spectra of compounds (7-12) in many respects are similar to those of 2-amino-substituted
pyrimidines [3,15] for which the bands of valence vibrations of the aminogroup
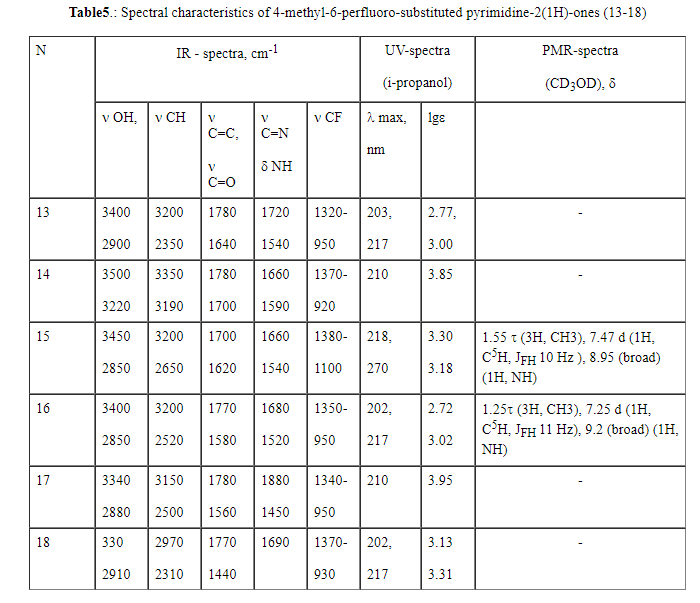
in a range of 3480-3330 cm-1 are typical. In the spectra of 2-hydroxypyrimidines
(13-18) an absorption band of 3500-3300 cm-1 corresponds to valence
vibrations of the hydroxyl group and a band of 1780-1770 is probably related
to the tautomeric form of hydroxypyrimidine-pyrimidine-2-one. The spectra
of 2-mercapto derivatives (19-24) contain a band of valence vibrations of
middle intensity of S-H group at 2600-2500 cm-1 and valence vibrations
of C-S at 665 cm-1. For all 6-perfluoro-substituted derivatives
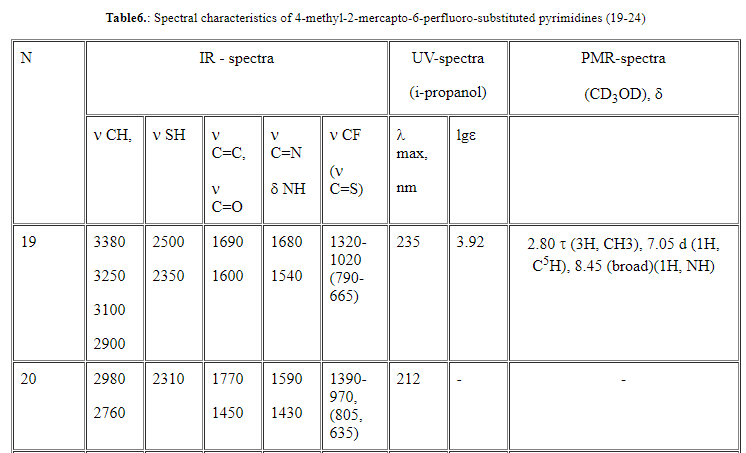
(7-24) ,valence vibrations of the methyl group in a range of 3220-2310 cm-1 , vibrations of the pyrimidine ring (  C=C, C=N, C=C, C=N, 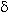 NH) at 1680-1440
cm-1 (skeletal vibrations of the cycle), 1340-900 cm-1 ( valence vibration of C-F bond) were recorded [20] (Tables 4-6). NH) at 1680-1440
cm-1 (skeletal vibrations of the cycle), 1340-900 cm-1 ( valence vibration of C-F bond) were recorded [20] (Tables 4-6).
The UV spectra of 6-perfluoro-substituted 2-aminopyrimidines (7-12) in a water-alcohol
solution exhibit one (compounds 7,8,10,11) or two absorption maxima (9,12)
associated with I 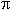 ->
* transition (compounds 7-12) in a range of 217-228nm (lg
->
* transition (compounds 7-12) in a range of 217-228nm (lg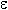 3.0404.15) and with II
->
* transition ( compounds 9,12) at 202-205nm (lg 3.98-3.05) which confirm the presence of multiple
bonds in the hetero ring [21] (Tables 4-6). In a strong acid medium (0.01M
HCl solution), electron spectra of the products (7,10) exhibit a hypsochromic
shift of the absorption bands by 22nm(7) and 17nm(10) with an absorption
maximum of 206nm (lg 3.23) and (lg 3.38). In an alkali medium (pH 14) for compounds (7,10)
a hypsochromic shift of the absorption bands was observed by 20nm (7) and
14nm(10), with a maximum at 208 nm (lg 2.72) and 209nm (lg 3.00). The isosbestic point (230nm)
at different values of pH is an evidence of the presence of two equilibrium
forms of the every investigated compounds (7,10)[22]. Two absorption bands
are observed in the spectra of compounds (13,15,16,18) recorded in aqueous
isopropanol at pH7: 202-218nm (lg 2.72-3.30) and a range of 217-270nm (lg 3.00-3.32) while compounds (14,17) have only
the first absorption band. An addition of a concentrated acid (0.01M HCl)
also leads to hypsochromic and hyperchromic shifts of the maximum absorption
band by 11 nm (lg 3.40 and 3.42) (13,16) at 206 nm. An addition
of an alkali (pH 14) also causes the hypsochromic shift of the maximum of
the absorption band by 8 nm at 209 nm. (lg 3.15 and lg 3.31) (13 and 16 respectively). At the wave length
of 230 nm and various values of pH similarly to the case of 2-aminopyrimidines
(7,10) , an isosbestic point also is observed allowing to consider such systems
(13,16) as two-component ones [22].The spectra of mercapto-derivatives of
pyrimidine (19,24) show two absorption maxima at 202nm (lg
2.74-3.14)
and 217nm (lg 3.01 and 3.35) (23,24), one maximum at 235nm
(lg 3.92-4.29) (19,21) and for compound (20) a maximum as
a shoulder at 212 nm ( Tables 4-6). 3.0404.15) and with II
->
* transition ( compounds 9,12) at 202-205nm (lg 3.98-3.05) which confirm the presence of multiple
bonds in the hetero ring [21] (Tables 4-6). In a strong acid medium (0.01M
HCl solution), electron spectra of the products (7,10) exhibit a hypsochromic
shift of the absorption bands by 22nm(7) and 17nm(10) with an absorption
maximum of 206nm (lg 3.23) and (lg 3.38). In an alkali medium (pH 14) for compounds (7,10)
a hypsochromic shift of the absorption bands was observed by 20nm (7) and
14nm(10), with a maximum at 208 nm (lg 2.72) and 209nm (lg 3.00). The isosbestic point (230nm)
at different values of pH is an evidence of the presence of two equilibrium
forms of the every investigated compounds (7,10)[22]. Two absorption bands
are observed in the spectra of compounds (13,15,16,18) recorded in aqueous
isopropanol at pH7: 202-218nm (lg 2.72-3.30) and a range of 217-270nm (lg 3.00-3.32) while compounds (14,17) have only
the first absorption band. An addition of a concentrated acid (0.01M HCl)
also leads to hypsochromic and hyperchromic shifts of the maximum absorption
band by 11 nm (lg 3.40 and 3.42) (13,16) at 206 nm. An addition
of an alkali (pH 14) also causes the hypsochromic shift of the maximum of
the absorption band by 8 nm at 209 nm. (lg 3.15 and lg 3.31) (13 and 16 respectively). At the wave length
of 230 nm and various values of pH similarly to the case of 2-aminopyrimidines
(7,10) , an isosbestic point also is observed allowing to consider such systems
(13,16) as two-component ones [22].The spectra of mercapto-derivatives of
pyrimidine (19,24) show two absorption maxima at 202nm (lg
2.74-3.14)
and 217nm (lg 3.01 and 3.35) (23,24), one maximum at 235nm
(lg 3.92-4.29) (19,21) and for compound (20) a maximum as
a shoulder at 212 nm ( Tables 4-6).
The PMR spectra of compounds (7, 9-11) in a range of 10.25-10.90 ppm. show a
broadened signal of NH protons pointing to the presence of two tautomeric
forms of pyrimidine derivatives : amine and imine ones. The spectra of products
(15,16) exhibit a broadened signal at 8.95-9.2 ppm assigned to the proton
bonded with the nitrogen of the pyrimidine ring, that also confirms the presence
of tautomeric form: pyrimidine-2-one. The spectra of pyrimidines (19,21,23)
recorded a broadened proton signal assigned to the cyclic nitrogen of the
ring in a range of 8.45-8.88 ppm. Moreover, for compounds (7, 9-11, 15, 16,19,21,23)
the proton signal at C5 at the pyrimidine ring as a singlet (7,10,19,21,23)
with a chemical shift of 6.90-7.47ppm and a doublet with (ppm.) are typical:7.00
JFH 50Hz (11), 7.47 JFH 34.4Hz (15), 7.70 JFH 50Hz and 7.75 JFH 34.4Hz ( Tables 4-6). The proton signal of the
S-H group at 4.75-4.92 ppm was observed in the spectra of compounds (19,21,23).
Integral intensities of the signals, their multiplicity and upfield chemicals
shifts (1.20-3.34 ppm) point to the presence of the methyl group in the pyrimidine
molecules.
The signals of fluorine atoms of the trifluoromethyl-2-undecafluoroxapentyl fragment
were observed in the 19F NMR spectra of 2-amino- (9) and 2-hydroxy-4-methyl-6-perfluoro(1-methyl-2-oxapentyl)
pyrimidine (15) in a range of 34-83.19 ppm ( relatively C6F6).
IR spectroscopy was used to evaluate thione-thiol equilibrium in ethanol
solutions of 2-mercapto-4-methyl-6-perfluorooctylpyrimidine (20)( 1 10-1 mol/l) in carbon tetrachloride ( 2.238) and in different mixtures of CCl4 and
CH3CN ( 36.02) [15], the thickness of the recorded layer was
1.08mm. The spectra of ethanol solutions of pyrimidine(20) in carbon tetrachloride
and also alcohol solutions of (20) in a mixture of carbon tetrachloride and
acetonitrile at different contents of the latter (20,60,90%) were analyzed.
Usually the vibration range of C=S group in heterocyclic systems coincides with
the range of deformation ring vibrations [20]. Based on the data of calculations
made for thiopyrimidines, we assume that the band of middle intensity at
1195-1205cm-1 is assigned to the vibration component
C=S + C-H
.
An absorption band at 1200cm-1 related to vibrations
C=S is observed in the spectra of alcohol solution of compound
(20) (Fig.2). Its intensity is minimal in CCl4 probably due to the presence
of thiol form (A).
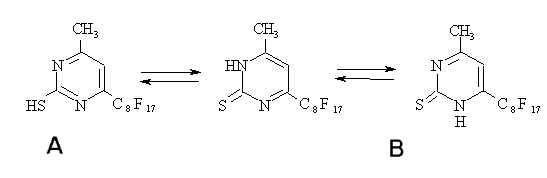
Along with an increase in polarity of the solvent ( in our case an increase in
the acetonitrile share) , the concentration of the tione form (B)
is increasing causing a growth of the band intensity at 1200 cm-1.
The effect of the solvent polarity on the ratio of tautomeric forms (A) and
(B) is determined by the differences in solvatation energies of heterocycle
-system [15]. Besides, at acetonitrile addition, a band in a
zone of 1180cm-1 appears that is associated with deformation ring
vibrations. The change in the heterocycle electronic structure at transition
from the thiol form to thione one leads to a significant decrease in its
intensity.
An effect of electron-deficient trifluoromethyl group in the 4 position on the
heteroring -system was reported in paper [15]. Probably, the
conjugation of lone-pair electrons of the sulfur atom with the aromatic system
is strengthened in comparison with the unfluorinated pyrimidine, that leads
to S=H bond strengthening. In contrast to the data of paper [15] , a significant
shift of the vibration band of 1230 cm-1 to the direction of high
frequences was not observed though its intensity increased. This band is
a summary one and includes vibrations C=S + C-H + NH.
The increase in the intensity may be explained by an increase
in the tione form (B) concentration (
C=S component). A negligible shift of this band at transition to a
more polar solvent depends on displacement
NH as a result of the formation of the hydrogen bond with proton-deficient
molecules of acetonitrile for example. In this case, absorption bands of
bond vibrations
C=S and
C-H appear as low-frequency shoulders on the new band of 1220 cm-1.(Fig. 2)
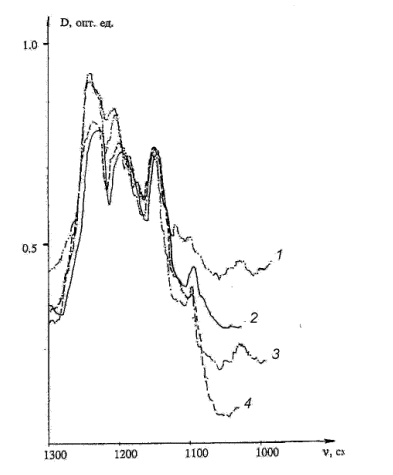
Fig. 2. IR spectra of the ethanol solution of pyrimidine (200 in the range of
1300….1100cm-1( in CCl4and its mixtures
with CH3CN). The thickness of the cuvette layer is 1.08.
Thus, the interaction of the asymmetric perfluoro-substituted
-diketones (1-6) with guanidinium carbonate, carbamide and thiocarbamide under
different conditions has been investigated. It has been shown that the appropriate
6-perfluoro-substituted 2-amino- (7-12), 2-hydroxy- (13-18) and 2-mercapto-
(19-24) 4-methylpyrimidines are formed as a result of the cyclocondensation
in alcohol or benzene ( in case of guanidine) in the presence of catalytic
amounts of hydrochloric acid.
Experimental.
IR spectra were recorded on a IKS-29 and a Shimadzu IR-470 (Japan) instrument
(film, KBr pellets).
UV spectra of aqueous and alcohol solutions of substances were performed on a
SF-26 spectrophotometer at a concentrations of the compounds of 10-4 mol/l, a thickness of the absorption layer of 1 cm.
PNR spectra of solutions of compounds in CD3OD were measured on a WM-250 (250MHz),
a Tesla-BS 487C (80 MHz) and a WF-200 (200 MHz) spectrometers, with GMDS as internal standard .
19F NMR spectra were recorded on a WF-200 (200MHz) instrument, with
C6F6 as external standard.
The control over the reaction course and purity of the compounds produced was
performed by TLC method on Sulifol UV-254 plates.
Perfluoro-substituted
-diketones (1-6) were synthesized by the condensation according to the method
[23,25].
2-amino-4 methyl-6-perfluorohexylpyrimidine
(7). 32.3 grams (0.08 mole) of 1,1,1,3,3-pentahydroperfluorodecane-2,4-dione (1) were added
to 7.04 grams (0.04 mole) of a suspension of guanidinium carbonate in isopropanol
and heated for 3 h. The solvent was distilled, a dry residue was dissolved
in acetone and transferred into water. The precipitated residue was filtered,
dried over P2O5.The yield was 17.0g (50%), colorless crystals, melting point
of 126-128oC ( from acetone).
2-amino-4-methyl-6-perfluorooctylpyrimidine (8), 2-amino-4-methyl-6-perfluoro(1-methyl-2-oxapentyl)
pyrimidine (9), 2-amino-4-methyl-6-perfluoro(1,4-dimethyl-2,5-dioxaoctyl)pyrimidine
(10), 2-amino-4-methyl-6-perfluoro(1,4,7-trimethyl-2,5,8-trioxaundecanyl)pyrimidine
(11) and 2-amino-4-methyl-6-perfluoro(1,4,7,10,13,16,19,22,25,28-decamethyl-2,5,8,11,14,17,20,23,26,29-decaoxaditriacontyl)pyrimidine
(12) were produced in much the same way as compound (7). The analytical and
spectral characteristics are given in
Tables 1 and 4.
4-methyl-6-perfluorohexylpyrimidine-2(1H)-one
(13). 15.0 g (0.04 mole) of 1,1,1,3,3-pentahydroperfluorodecane-2,4-dione (1) were added
to a mixture of 2.23g (0.04 mole) of carbamide in isopropanol and 1 ml of
HCl. The reaction mass was kept at 80-100oC for 3-5 hours. The
solvent was distilled, the dry residue was sublimated under vacuum. The yield
was 10.0g (68%), colorless crystals, melting point of 98-100oC.
4-methyl-6-perfluorooctylpyrimidine-2(1H)-one (14), 4-methyl-6-perfluoro (1-methyl-2-oxapentyl)pyrimidine-2(1H)-one
(15), 4-methyl-6-perfluoro(1,4 -dimethyl-2,5-dioxaoctyl)-pyrimidine-2(1H)-one
(16), 4-methyl-6-perfluoro(1,4,7-trimethyl-2,5,8-trioxaundecanyl)-pyrimidine-2(1H)-one
(17) and 4-methyl-6-perfluoro (1,4,7,10,13,16,19,22,25,28-decamethyl-2,5,8,11,14,17,20,23,26,29-decaoxaditriacontyl)pyrimidine-2(1H0-one
(18) were produced in much the same way as(13). The analytical and spectral
data are given in Tables 2 and 5.
4-methyl-2-mercapto-6-perfluorohexylpyrimidine
(19). 28.28g (0.07 mole) of 1,1,1,3,3-pentahydroperfluorodecane-2,4-dione (1) were added
to 6.08g (0.08 mole) of a solution of thiocarbamide in isopropanol and 2
ml of concentrated HCl. The reaction mass was kept at 70oC for
10-12h. The oily product was extracted, dried, the solvent was distilled,
the product was recrystallized from acetone. The yield was 33.0g (60%), pink
crystals, melting point of 114-116oC.
Similarly the following substances were produced: 4-methyl-2mercapto-6-perfluorooctylpyrimidine
(20), 4-methyl-2-mercapto-6-perfluoro(1-methyl-2-oxapentyl)pyrimidine (21),
4-methyl-2-mercapto-6-perfluoro (1,4-dimethyl-2,5-dioxaoctyl)pyrimidine (22),
4-methyl-2-mercapto-6-perfluoro(1,4,7-trimethyl-2,5,8-trioxaundecanyl)pyrimidine
(23) and 4-methyl-2-mercapto-6-perfluoro(1,4,7,10,13, 16,19,22,25,29-decamethyl-2,5,8,11,14,17,20,23,26,29-decaoxaditriacontyl)
pyrimidine (24). The analytic and spectral data are given in Tables 3 and 6.
References
1. Brown D.J. The Pyrimidines. - N.Y.- London: Intersci. publ., 1962. - 744 p.
2. Brown D.J. The Pyrimidines. Suppl.I. N.Y.- London: Intersci. publ., 1970.
- 899 p.
3. Brown D.J. The Pyrimidines. Suppl. II. N.Y.-London: Intersci. publ., 1985.
- 916 p.
4. Nishiwaki T. // Bull.Chem.Soc.Jap. - 1970. - V. 43, N 3. - P. 937-939.
5. Kreutzberger A., Leyke-Rohling S. // J.Heterocycl.Chem. - 1978. - V. 15. -
C. 1097-1099.
6. Kreutzberger A., Leyke-Rohling S. // J.Fluorine Chem. - 1979. - V. 14, N 2.
- P. 55-63.
7. Kreutzberger A. // J.Fluorine Chem. - 1979. - V. 14, N 2. - P. 131-138.
8. Kreutzberger A., Schimmelpfenning H. // J.Fluorine Chem.- 1980. - V. 15, N
6. - P. 511-517.
9. Kreutzberger A., Sellheim M. // J.Fluorine Chem. - 1985. - V. 27, N 2. - P.
203-212.
10. Kreutzberger A., Gillessen J. // J. Fluorine Chem. - 1985.- V.29, N 3-4.-
P. 385-397.
11. Kreutzberger A., Burger A. // J.Fluorine Chem. - 1993. - V. 60, N 2-3. -
P. 257-261.
12. Pathak V.N., Sareen V., Joshi K.C. // J.Fluorine Chem. - 1985. - V. 29, N
1-2. - P. 202.
13. Kucerovy A, Mattner P.G., Hathaway J.S., Repic O. // Synth.Commun. - 1990.
- V. 20, N 6. - P. 913-917.
14. Butter A.R., Leitch E. // J.Chem.Soc., Perkin Trans.II, 1976. - P. 832-835.
15. Gerus I.I., Vdovenko S.I., Gorbunova M.G., Kuhar' V.P. // KhGS. - 1991. -
N 4. - C. 502-511.
16. Pashkevich K.I., Saloutin V.I., Postovskij I.YA. // Uspekhi khimii.- 1981.-
T.50.- Vyp. 2. - S. 325-354.
17. Allen G., Dwek R.A. // J.Chem.Soc.(B). - 1966. - N 1. P. 161-163.
18. Saiks P. Mekhanizmy reaktsij v organicheskoj khimii. - M.: Khimiya, 1991.
- 448 s.
19. Knorre D.G., Krylova L.F., Muzykantov V.S. Fizicheskaya khimiya. - M.: Vysshaya
shkola, 1990. - S. 372-375.
20. Bellami L. Infrakrasnye spektry slozhnyh molekul. - M.: IL, 1963. - 506 s.
21. Kazitsyna L.A., Kupletskaya N.B. Primenenie UF-, IR- i NMR-spektroskopii
v organicheskoj khimii. - M.: Vysshaya shkola, 1971. - 264 s.
22. Bernshtejn I.Ya., Kaminskij Yu.L. Spektrofotometricheskij analiz v organicheskoj
khimii. - L.: Khimiya, 1986. - 200 s.
23. Peshkova V.M., Mel'chakova N.V. -Diketony. - M.: Nauka, 1986.- 200 s.
24. Trishina A.Yu., Irisova E.V., Popova L.M., Vershilov S.V., Zachinyaev Ya.V.,
Maksimov B.N., Ginak A.I. // Khim. i tekhnol. organich. soed. sery: Tez.
dokl. Vseross. konf., Kazan', 1995 g. - S. 173.
25. Trishina A.Yu., Popova L.M., Irisova E.V., Vershilov S.V., Konyuhova S.V.,
Ginak A.I. // Tez. dokl. mezhinstitut. Kollokviuma "Khimiya azotistyh geterotsiklov"
Chernogolovka, 18 oktyabrya 1995 g. - S. 96.
|