Fluorine Notes, 1998, 1, 1-2
Tetrafluoroethylene oxide and derivatives on its base
V. Andriushin and N. Pavlova
Part 1.Synthesis methods of tetrafluoroethylene
INTRODUCTION
Synthesis methods
1. Photochemical oxidation of tetrafluoroethylene
2. Radiochemical oxidation of tetrafluoroethylene
3. High temperature oxidation of tetrafluoroethylene
4. Catalytic methods of tetrafluoroethylene oxide synthesis
CONCLUSIONS
LITERATURE CITED
INTRODUCTION
In the recent years an increased interest to chemistry and technology of perfluoro-olefin oxides was caused by their high reactivity and variety of oligomers and polymer materials produced on their base possessing unique physico-mechanical properties and exploitation characteristics which allowed to solve very different problems in nuclear power engineering, radioelectronics, space and aircraft technique and other newest branches of science and technique (1,2).
Such materials possess resistance to environmental unfavorable factors and aggressive chemicals with preservation of their own physico-chemical properties over a wide temperature range. Polymers based on tetrafluoro-ethylene oxide (TFEO) are of particular interest. Among them there are crystal polytetrafluoroethylene oxide surpassing traditional Teflon by a number of indices, fluororubbers with a glass transition temperature to minus 50-minus 80oC produced by co-polymerization of vinylidene fluoride and perfluoroalkylvinyl ethers on the basis of TFEO, membrane materials, lubricants, surfactants and many other substances produced from bifunctional fluoromonomers based on TFEO and difluoroanhydrides of perfluoro-dicarbonic acids (3-12).
This paper is an attempt to systematize data on TFEO properties available in literature and to draw attention of researchers to wide possibilities to create unique materials on its base
METHODS of TFEO SYNTHESIS
Despite the fact that several groups of researchers from different countries
( mainly Italy, the USA, the USSR) have been working intensively on the development of synthesis methods and investigation of TFEO chemical transformations , up to now there are no data on methods of industrial TFEO synthesis because (as we believe) of hazard of such a production caused by TFEO properties.
First TFEO syntheses were performed in the early 1960s (13-23). The most of them are based on a reaction of tetrafluoroethylene (TFE) oxidation by molecular oxygen by exposing to different initiators. They are classified conditionally into several groups mentioned below just according to the methods of initiation.
1. Photochemical tetrafluoroethylene oxidation.
Tetrafluoroethylene oxidation by oxygen at exposure to mercury-quartz lamps of 1800-3200 A wave length is technologically arranged in different ways: either in gas phase flow-type reactors or in a medium of inert solvents with or without use of different photostabilizers (photoinhibitors)in the both types of the reactors (14, 24-29).
According to patent (24) of France the gas phase photochemical TFE oxidation by elemental oxygen ( at a temperature of 30-150oC, the pressure of 2 atm, ratio of TFE / O2 = 1-16/4, contact time of 8-10 minutes) has TFEO yield equal to 40-52% regards TFE reacted, which conversion comes up to 56-66%. This process is obviously low productive ( the time of gas contact is great) and of low efficiency ( the yield is only about 50%) with a complex system of goal product purification from gas admixtures such as carbonyl fluoride, perfluorocyclopropane, carbon dioxide; liquid and solid reaction products of perfluoropolyoxyalkylene structure.
The reaction equation may be represented by the following scheme:
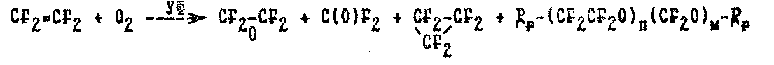
The method of gas phase photochemical TFE oxidation in the presence of sensitizers such as bromine, chlorine, iodine, nitrogen oxide and dioxide, hydrogen bromide (14,26) is more efficient. Bromine is the most active among the listed sensitizers. Gas phase photochemical oxidation is extremely sensible to the amount of the sensitizer added: the optimal ratio of the bromine amount to the total flow is 40-60 parts to 1 000 000.
The gas phase TFE oxidation in the presence of bromine at the ratio TFE/O2=1.25 and a temperature of 130-150oC has TFEO yield equal to 21-23% regards TFE reacted with TFE conversion of 65-70%. At the same time the residence time of reagents in a reaction area is 1.5 minute. To decrease a danger of explosion for the gas phase process, oxygen was diluted with inert gas (nitrogen or helium). The same process (14,25,26) carried out in a medium of inert liquid allowed to increase slightly reaction selectivity ( the yield of TFEO increased up to 45-46% at the same conversion of TFE). Besides, the use of an inert solvent assumes a more wide concentration range of a sensitizer added without significant deviations from the maximum value of TFEO yield. The formation of epoxide under bromine exposure occurs even in UV radiation absence, but the yield is ca.10 times less .
Liquid (at the reaction temperature) saturated perhalogenated hydrocar-bons and ethers were used as inert solvents .
The process carried out in liqud PEF-180 ( which is a product of hexafluoropropylene photooxidation polymerization , b.p.=180oC) with use of photo-sensitizers ( bromine) and reaction initiators containing a movable haloid atom ( bromine halons, for example) at 70-115oC is characterized by a high yield of TFEO ( up to 82%) and TFE conversion to 66%. In this case initiators may be added just as independent solvents so also as small additions (0.5-5%) to other solvents. In case of their absence the conversion drops to 15-35% and yield to 65-67%. Hexafluoroazomethane may be used also as an effective initiator of photochemical TFE oxidation (15, 16, 29).
The mechanism of photochemical oxidation of terminal fluoro-olefins by molecular oxygen both in gas and liquid phases has been discussed by many authors (20,21, 30-34) and at present there is no doubts that TFE oxidation proceeds according to a free-radical chain mechanism. This mechanism includes many elementary stages of continuation and termination of chains, also stages of decomposition of free radicals resulting in the formation of nonradical products . That is why the composition of the products and the mechanism of oxidation change over a wide range in dependence on the process conditions.
According to up-to date concepts the mechanism of this reaction may be given by the scheme considering TFE oxidation by oxygen as copolymerization of TFE molecules and oxygen with simultaneous decomposition of the growing radicals across bonds O-O and C-C:
|
initiator |
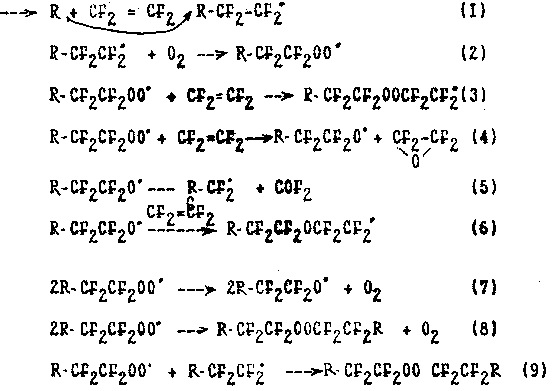
When the temperature of initiated TFE oxidation is increasing the stages of decomposition of the growing radicals (4), (5) become pevailing due to more high activation energy than for stages (2), (3). As a consequence of this there is observed a yield decrease of polymer products of CF3O-(CF2CF2O)n-(CF2CF2 OO)m-(CF2O)p-CF2OF structure, a decrease in molecular mass, the prevailing of fragments of difluoromethylenoxide structure in the polymer,an increase in yield of TFEO and carbonylfluoride . At a temperature above 200oC the products of double bond termination become practically the sole.
Based on the kinetic investigation of the low-temperature HFP oxidation in gas phase a conclusion was made that the rates of epoxidation in comparison with the rates of forming carbonyl-containing products depend on the olefin concentration largely and to a lesser degree depend on the initiation rate. Consequently the selectivity of epoxidation in the processes of radical oxidation of fluoro-olefins increases when the olefin concentration is growing and the initiation rate is decreasing (30-35).
The mechanism of photochemical TFE oxidation by oxygen in the presence of mercury vapors has been suggested by Heicklin J. et al. (25, 36, 37). In conformance with this mechanism, the energy of UV radiation turns into the energy of electronic excitation of sensitizer with the further formation of the peroxide difluorocarbene radical (.CF2OO.), also singlet (..CF21) and triplet (..CF23) radicals of difluorocarbene according to the following scheme:
Hg ->Hg*
Hg* + O2 -> Hg + O2*
O2* + C2F4->C2F4* + O2
C2F4* + O2->.CF2OO. + :CF2I
.CF2OO.+ :CF2I ->2COF2
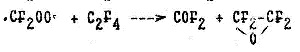
.CF2OO.+ C2F4-> 2COF2+ :CF23
:CF23+ O2-> .CF2OO.
2.CF2OO. -> 2COF2+ O2
The sensitizing action of bromine appears due to its dissociation into radicals under UV exposure. Atomic bromine adjoining the olefine molecules initiates TFE oxidation:
Br2 ->2Br*
Br* + C2F4 -> BrCF2CF2*
BrCF2CF2* + O2 ->BrCF2CF2OO*
and further in much the same way as stages (3)-(9).
Some examples given above on the establishment of photochemical TFE oxidation by oxygen ,particularly using initiators and solvents, could be the basis for the development of TFEO production. But such obstacles as equipment complexity, danger and high power capacity were not overcame in full and up to now the photochemical method is used only in laboratory practice.
- 2. Radiochemical tetrafluoroethylene oxidation
The synthesis of TFEO by the radiochemical method is similar to the described above photochemical TFE oxidation by elemental oxygen with the only difference that initiation of the chain-radical reaction mechanism is performed by different sources of ionizing radiation. The process mechanism is quite described by reactions (1)-(9).
As a rule, radioactive Co60 (0.4 milliroentgen/hour) serves as a source of g -radiation and a roentgen device(1.55 milliroentgen/hour) is used as a source of X-rays.
The influence of some technological factorson the process of radiochemical TFE oxidation has been tested in papers (3,14,18). So, at room temperature and atmospheric pressure in gas phase at the equimolar ratio of the reagents , carbonylfluoride (CBF) and liquid olygomers(polymers) of peroxide structure are formed in addition to the goal TFEO. The decrease in temperature from +25 to -25oC is resulting in a decrease in olefin conversion and relative yield of gas products, at the same time the ratio TFEO/CBF remains practically the same. An increase in TFE partial pressure in the initial gas mixture causes a substantial increase in TFE epoxidation rate, at the same time a change in the oxygen pressure does not influence the reaction rate.
The oxidation of TFE with oxygen at gamma-quantum initiation at an elevated pressure allows to increase significantly yield of the goal reaction products (38-40), for example the oxidation of TFE by oxygen under exposure to gamma-rays (Co60) of intensity of 2.7*105 roent/hour at 15oC and a pressure of 7-9.5 atm makes possible to produce TFEO with the yield of about 50% and TFE conversion of 60-70% (39).
It has been shown in the work of Poluektov et al. (41,42) that the yield of TFEO is increased with a decrease in initiation rate of the chain-radical TFE oxidation by means of the reaction temperature reduction. So, at the homogeneous gas phase TFE oxidation initiated with gamma-quanta within the temperature range of minus 60 to plus 37oC , it was possible to produce the oxide with the yield of 50%. It was determined that at temperatures below minus 3oC the reaction rate was increasing with a temperature decrease (41).
An improved method of radiochemical TFE epoxidation is gas phase TFE oxidation by oxygen in flow at gamma-quantum initiation in the presence of higher perfluoro-olefins (produced by polytetrafluoroethylene radiothermal cracking ) which absorbing gamma-quantum energy form macroradicals initiating the reaction. The oxidation of TFE at atmospheric pressure in the presence of polytetrafluoroethylene cuttings under exposure to gamma-quanta from Co60 ( with a capacity of doses absorbed of 0.08-1.4 J/kg, within the temperature range of -50 - +125oC, at the mole ratio of TFE/O2= from 9/1 to 1/9, the contact time of the reagents varies from 1 to 12 min.) gives the yield of TFEO up to 75-76% regards TFE reacted at 80% conversion of TFE . Irradiation termination results in a sharp drop of the reaction rate. The increase in rate of radiochemical oxidation in the presence of catalyst in comparison with the homogeneous TFE oxidation is explained by the increase in reaction initiation rate in the presence of the irradiated polytetrafluoroethylene because its density is more than that for the gas mixture by a factor of 103 , consequently the radiation absorption is more by 3 orders of magnitude. Furthermore , the solid surface of the catalyst promotes scattering the energy of an excited oxide molecule that results in increasing selectivity of the oxidation reaction and increase in TFEO yield (42).
The reaction of TFE oxidation by air oxygen in the presence of Fluoroplast-40 powder preliminarily irradiated by gamma-rays of Co60 and containing 10 18gr-1 of free radicals has been studied in paper (42). The best results on TFEO yield were obtained within the temperature range of 56-93oC where the conversion of TFE was about 80% and TFEO yield reached 70-75% regards TFE reacted. Gradually the conversion of TFE is decreasing and when the concentration of free radicals becomes 1/3 of the initial value ( after 1.5-2 hours) the oxidation is terminated practically. There are up to some hundred of TFE molecules entered the oxidation reaction for every perished radical.
At contact with air oxygen all the free radicals of the irradiated Fluoroplast quickly transfer from the alkyl form to the peroxide one. Just as fluoroalkyl and peroxide radicals are rigidly fixed in the fluoroplast structure and are able to recombine only at direct contact so the only primary reaction is considered: the heterogeneous process of chain growth (43)
Rf OO* + CF2=CF2 --> RfOOCF2CF2* (10),
where Rf is the fluoroplast polymethylene chain.
Further the process proceeds by the usual mechanism of olefin halides oxidation:
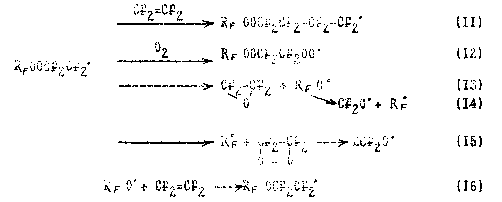
The relation of the rates of stages (11)-(15) depends on the experimental conditions. At low temperatures of -50 - -70oC monomolecular stages of decomposition (13), (14), and (15) are retarded and the process mainly is governed by bimolecular stages of polymer chain growth (11) and (12). At increase in temperature the share of the gas phase oxidation products is rised , this points to the increase in reaction rates (13), (14), (15).
During the process of chaingrowth and destruction the radical active center changes its location in the Fluoroplast structure. In this case a possibility to meet active centers arises resulting in recombination of appropriate radicals and is a reason for reduction in oxidation reaction rate after irradiation is terminated.
This scheme of the oxidation process arrangement is of practical interest due to the high selectivity of the epoxidation reaction, but high doses of irradiation and a comparatively short time of initiator action (1.5-2 hours) put some technological difficulties in scaling up the reactor unit to industrial one.
At the same time the Fluoroplast after repeated intensive ionizing irradiation loses its mechanical strength due to destruction connected with detachment of the difluoromethylene fragments from the initial radical ( see reaction 14).
- 3. High temperature tetrafluoroethylene oxidation.
Apart from the mentioned above methods, initiation of the reaction of TFE oxidation by oxygen may be done by subjection of the reaction mixture to heat.
Patent (44) has described TFEO production at the temperature of 100oC and pressure of 20 atm with 63% yield of TFEO at TFE conversion of up to 90% with the formation of perfluorocyclopropane and carbonylfluoride as by-products. The process is dangerously explosive. To avoid TFEO isomeriza-tion and to reduce process danger, it is carried out in a medium of inert solvent. To support the necessary olefin concentration in the liquid phase a high pressure is required, so the process is carried out in autoclaves of alloys of copper, nickel anf chromine, titanium, silver, platinum. But even in this case the process remains hazardous to be used on industrial scale.
4. Catalytic methods TFEO synthesis.
This group of the methods of TFEO synthesis unites the processes of TFEO oxidation by oxygen initiated by various chemical reagents able to form epoxidation agents under the process conditions.
These are:
- ozone, fluorine, oxygen difluoride, peroxydisulfuryl difluoride ( homoge-neous catalysis);
- metal silver, silver oxides, also mixtures based on silicon dioxide and silver oxides with oxides and salts of other metals ( heterogeneous catalysis);
- potassium permanganate solution in anhydrous hydrogen fluoride (ionic TFE oxidation).
Great Britain patent (49) recommends to carry out the gas phase TFE oxidation by ozonized oxygen in a flow-type reactor at a temperature of 0-40oC at the ozone content of ca. 1.6% mol.( the content of ozone in ozonized oxygen is ca. 3%). In the equimolar mixture of TFE and oxygen the yield of TFEO reachs 46% at 41.3% conversion. Carbonyl fluoride and liquid polymers of polyoxaperfluoro-methylene structure with a molecular mass of about 1500 are formed as reaction by-products .
A significant increase in selectivity of epoxidation reaction is obtained when the process is carried out in a solvent. According to patent (50) of France , TFEO yield is 62% and TFE conversion is 48.8% when twofold excess of TFE is oxidized by ozonized oxygen ( the ozone content is 2%) in R-113 at the temperature of 15oC and atmospheric pressure . Moreover, it was produced 15.8% of polymer of the same structure with a molecular mass of about 8000. The mechanism of the reaction of TFE oxidation by ozone was studied by many authors (17, 51-58) and according to modern concepts includes the following stages:
C2F4 +O3 -> (C2F4O3) -> CF2O + CF2OO* (17)
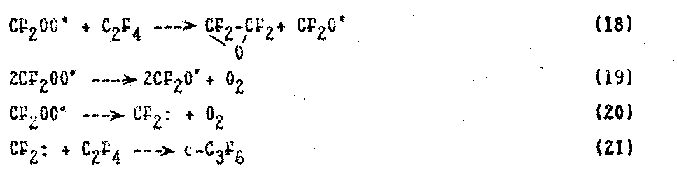
The mechanism of reaction (17) with formation of intermediate ozonide and its conversion to carbonyl fluoride and difluorocarbonyloxide has been considered in details in papers (56, 58) and is given by the following scheme:
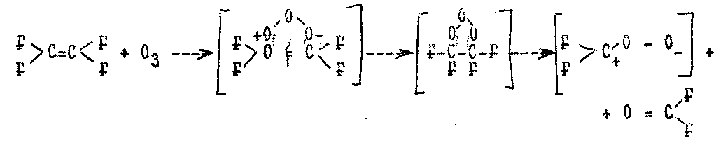
Authors (30,31,54) believe that the mechanism of TFE oxidation by oxygen in the presence of ozone represents addition of olefin and oxygen by turn to the growing radical with consequent detachment of the volatile products: TFEO and carbonyl fluoride . Reaction initiation is performed to(before) reaction (17) with biradical CF2OO* isomerization to *OCF2O ; its continuation is described upon the whole by the equations of reactions (1)-(9).
Oxygen difluoride is an effective initiator of TFE oxidation by molecular oxygen (45-48). For example, in patent (46) of the USA TFEO yield reached 81% with TFE conversion of 87%. Oxidation was carried out at room temperature and atmospheric pressure, the contact time was 3 hours and the content of oxygen difluoride in oxidezer was appr. 3.8% mol. To reduce explosion hazard of the process, the reactions are carried out in a medium of an inert tetrafluoromethane (carbon fluoride) liquid . Producing TFEO with such a high selectivity is in conflict with the oxidation mechanism proposed by authors (30,31) according to which the selectivity of epoxidation should not exceed 67% by volume.
To explain the high selectivity of fluoro-olefins epoxidation and to reduce the selectivity of the process at increase in oxygen concentration, it was proposed to complete the scheme of oxidation (1)-(9) with the stage of decomposition of perfluoroperoxyalkyl radicals into two oxide molecules (33,59-62):
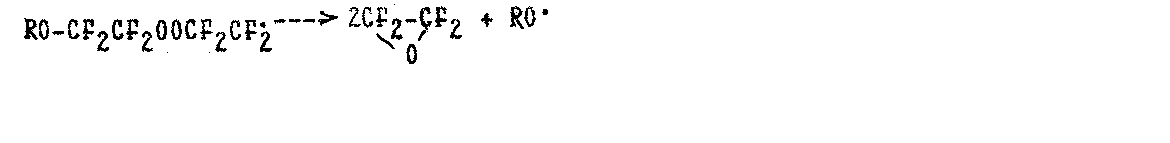
and also the stage of isomerization and decomposition of perfluoroalkyl-peroxi radicals with interruptionof the carbon skeleton (61):
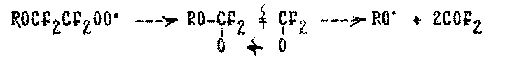
Peroxidisulfuryldifluoride evidently acts similarly to the described above catalysis by oxygen difluoride. The authors of the present paper ( with A.V.Veber participation) have determined that the reaction of TFE oxidation by oxygen in the presence of peroxidisulfuryldifluoride in gas phaseis highly exothermic , while various packings ( copper , nickel, bronze, iron) are inefficient : as a result there were separated only fluoroanhydryde of trifluoroacetic acid and a,w -bis ( fluorosulfonyloxi) polytetrafluoroethylene.
The reaction was successful only in a medium of inert fluorocarbons with use of Fluoroplast-4 packing :the yield of TFEO was 65-76% with conversion of TFE of 65-72% at -5- 60oC. With regard to using trifluoromethylhypo- fluorite as an initiator of the reaction of TFE oxidation, its function is explained by CF3OF diccosiation across bond O-F and further reaction with TFE and oxygen according to the free-radical mechanism (71).
Catalytic heterogeneous reactions regards conditions ,chemical nature of oxidezers used and character of reaction products take an intermediate place between the processes of ionic and radical oxidation of fluoro-olefins. Catalyst is to be activated by heating in oxygen flow to carry out the process successfully.
The oxidation of TFE by oxygen on granular argentic oxide (activated at 120oC for 50 hours) in a flow-type reactor at the temperature of 120oC, atmospheric pressure and threefold oxygen excess gives the yield of TFEO of 57% with TFE conversion of 26.6%. The lifetime of the catalyst is 200 hours (63). The conversion of olefin can be increased to 35-70% with TFEO yield of 55.4-64.5 % if the temperature of catalyst activation is increased to 400-550oC and the oxidation is carried out at 150-160oC.
The mechanism of TFE oxidation by oxygen on silver catalyst is similar to the mechasim of ethylene epoxidation (33,66,67). Just as TFE is significantly more active than ethylene in reactions with nucleophilic agents (68) so it is makes easier its interaction with the oxygen chemisorbed on the silver surface in form of peroxide anion O2- which is an epoxidation agent (69). Catalyst fouling is explained by the formation of silver fluorides on its surface due to its interaction with the reaction by-product : carbonylfluoride (66).
TFE oxidation on silica gel catalyst requires preliminary activation of the catalyst with a mixture of oxygen and olefin at a temperature of 240-200oC for 1-20 hours. TFEO yield attains 70% at TFE conversion of 72% (65). The catalyst requires periodical regeneration because accumulation of liquid polymer products of peroxide structure decreases strongly its activity.
An interesting process from chemical point of view is TFE oxidation by potassium permanganate in anhydrous hydrogen fluoride (70) which is represented by authors as reactions of electrophilic addition, while fluoroanhydride of manganic acid( which is forming at the reaction of potassium permanganate with hydrogen fluoride) acts as an electrophilic agent:
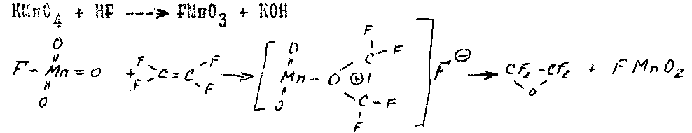
The reaction was carried out at the temperature of -70oC and there was produced a mixture of TFEO and fluoroanhydride of trifluoroacetic acid.
CONCLUSIONS
The given analysis of the well-known methods of TFEO production has shown that each of them possesses a number of substantial disadvantages preventing their application for technological process establishment. Thus, the photochemical methods of TFE oxidation require a powerful source of irradiation resulting in significant energy consumption at low productivity of reactors. The catalytic heterogenous processes are distinguished by rapid catalyst deactivation.The radiochemical synthesis methods are characterized by high doses of irradiation and equipment complexity. The oxidation of TFE on polytetrafluoroethylene powder requires frequent repeated irradiation of the Fluoroplast resulting in its mechanical destruction. The thermal TFE oxidation under pressure and also its oxidation in the presence of oxygen difluoride are connected with significant explosive danger. The majority of the well-known methods do not possess sufficient selectivity: along with TFEO a big quantity of liquid and gas by-products are produced, also there are low factors of TFE conversion and TFEO yield.
Namely these factors and total explosive danger of TFEO producing determine the absence of tetrafluoroethylene industrial production up to the present time.
The particular choice of the method for scientific research and for production of small samples of TFEO is determined by specific capabilities of Research Centres and their traditions. According to our reckoning the most interest to establish a pilot production or pilot installations is in TFE oxidation by molecular oxygen in the presence of chemical initiators ( chlorine, bromine, ozone, trifluoromethylhypofluorite, peroxidisulfuryldifluoride). This method is enough simple in equipment design, able to provide high productivity in a flow-type reactor at moderate temperatures.
LITERATURE CITED
2. Novoe v tekhnologii soedinenij ftora (Pod red. N. Isikawa)- M.; Mir, 1984, s. 391-393.
3. Donato A., Lenzi M., Mele A. Thermal decomposition of polytetrafluoroetylene oxide.-J.Macromol. Sci.-Chem., 1967, A1 N 3, p.429-438.
4. Eleuterio H.S., Polimerisation of perfluoro epoxides - J.Macromol. Sci-Chem., 1972, A6, N 6, p. 1027-1052.
5. Warnell J.L.,US Patent N 3449389
6. Fritz C.G., Warnell J.L., US Patent N 3317484.
7. Warnell J.L., US Patent N 3250806.
8. Evers R.C.,US Patent N 4147858
9. Baucom K.,US Patent N 4241223.
10. Evers R.C.,US Patent N 4108884.
11. Patentappl.JP 54-135722 от 22.10.79.
12. Patentappl.JP54-135723 от 22.10.79.
13. Tomanovskaya V.F. Okisi tetraftorehtilena, geksaftorpropilena, perftortsiklogeksena - Obzor zhurnal'noj i patentnoj literatury. 1969-1973, L., 1974.
14. Warnell J.L., Patent UK N 931587, 1963.
15. Ginzburg V.A., Vlasova E.S., Vasil'eva M.I. i dr., Fotoreaktsii geksaftorazometana s nepredel'nymi soedineniyami.-Dokl. AN SSSR, 1963, t.149, s. 97.
16. Ginzburg V.A., Vasil'eva M.I., Poluchenie i nekotorye khimicheskie svoistva okisi tetraftorehtilena.-ZhOKh, 1967, t.37, N 11, s. 2493.
17. Gozzo F., Camaggi G., Oxidation reaction of TFE and their products.I, Auto-oxidation.-Teterahedron, 1966,V. 22, N6, p.1765-1770.
18. Caglioti V., Lenzi M., Mele A., Radiation-induced oxidation of tetrafluoroethylene.-Nature,1964,V. 201,N 4919, p.610-611.
19. Caglioti V., Delle Site A., Lenzi M., Mele A., Oxidation of TFE by Molecular Oxygen- J. Chem. Soc., 1964, V.12, p. 5430-5433.
20. Cordischi D.,Lenzi M., Mele A., Kinetics of the oxydation of tetrafluoroethylene by molecular oxygene- Trans. Farad. Soc., 1964, V.60, N 11, p.2047-2053.
21. Gozzo F., Camaggi G., Oxidation reactions of tetrafluoroethylene and products there from. II. Photochemical reactions.- Tetrahedron, 1966, V.22, N 7, p. 2181-2190.
22. Tarrant P., Stamp E. Okisi perftorolefinov- ZhVKhO im. D.I. Mendeleeva, 1970, t.15, vyp. 1, s. 34-44.
23. Tarrant P., Allison C.G., Bartold K.P. Fluorine-containing epoxides.-Fluorine Chemistry Reviews, 1971, 5, p. 77-112.
24. Societa Edison S.p.a., Pat. France N 1499859,1967. 25. Heicklen J., Knight V.A. Reexamination of the mercuryphotosensibilized oxidation of TFE.- J. Phys. Chem.,1966,V.70, N 12, p.3901.
26. Biggs H.H., Warnell J.L., Pat. France N 13225997, 1963
27. Du Pont. Details chemistry of perfluorocarbone epoxides.- Chem.Eng.News, 1967, V. 45, N 33, p.18
28. Sianesi D., Pasetti A., Belazdinelle G.- Pat. France N 1574703, 1970.
29. Vilenchik Ya.M., Mitrofanova L.N., Senichev Yu.N. Fotohimicheskij sintez okisi tetraftorehtilena.- ZhOrKh, 1978, t. 14, vyp.8, s. 1587.
30. Sianesi D., Pasetti A., Bernardi C., Caporiccio G., Synthesis of perfluoropolyethers by photooxidation of fluoroolefines.-Chem. ind., 1973, V.55, p.208.
31. Mayo F.R., Miller A.A., Russel G. A., The oxidation of unsaturated compounds.- J. Am.Chem. Soc., 1958, V.80, p.2497.
32. Poluektov V.A., Chernyavskij A.I. Kinetika i mekhanizm okisleniya galoidolefinov.- Neftekhimiya, 1978, t. 18, N 4, s. 558-564.
33. Sokolov L.F., Valov P.I., Sokolov S.V. Poluchenie monomerov dlya sinteza ftoruglerodnyh polimerov okisleniem ftorolefinov.-Uspekhikhimii. 1984, t. 53, N 7, s. 1222-1246.
34. Advances in Photochemistry. Heicklen J. Gas phase oxidation of perhalocarbons.-1969, 7, p.58-147.
35. Ponomarenko V.A., Krukovskij S.P., Alybina A.Yu. Ftorsoderzhashchie geterotsepnye polimery.- M., Nauka, 1973, S. 50-146.
36. Heicklen J., Knight V., Reactions of oxygen atoms with tetrafluoroethylene in the presence of molecular oxygen.-J. Phys. Chem., 1966, V.70, N 12, p. 3893
37. Saunders D., Heicklen J., Some reactions of oxygen atoms.- J.Phys. Chem., 1966,V.70,N 6,p.1950.
38. Higani Hiromiti, Tabata Jon Ekho i dr. Radiatsionnoe okislenie tetraftorehtilena.- Koge Kaganu dzami, 1969, t. 72, N 8, s. 1884-1886- C.A., 1970, 17217K.
39. Patent appl. JP 48-19610, 17.07.68.
40.Patentappl.JP73-19610, 14.06.73.
41. Poluektov V.A., Begishev I.R., Mezentsev A.I., Shapovalov V.V., Izmenenie kineticheskih parametrov tsepnoj reaktsii pri uchastii v ehlementarnyh aktah donorno-aktseptornyh kompleksov aktivnyh chastits s molekulami reagentov. -Dokl. AN SSSR, 1979 , t.245, N 1, s. 148.
42. Mezentsev A.I., Poluektov V.A. Issledovanie kinetiki gazofaznogo initsiirovaniya gamma-kvantami okisleniya TFEH v prisutstvii katalizatorov initsiatorov vysshih perftorolefinov.-ZhFKh, 1983, t. 57, vyp.7, s. 1672.
43. Markevich A.M., Klejmenov N.A., Volohovich I.E., Mel'nikov V.P., Soklov S.V. i dr. Okislenie TFEH kislorodom vozduha na poroshkoobraznom politetraftorehtilene, soderzhashchem svobodnye radikaly.-Kinetika i kataliz, 1979, t.20, vyp.3, s. 785-788.
44. CarlsonD.P., Pat.US 3576733.
45. Ruff J.K., Merritt R.F. Oxidation with oxygen difluoride-J.Org. Chem., 1965, V.30, N 11, p. 3968-70.
46. Winmayr V., Pat. US N 3639429.
47. Dale J.W., Pat US N 3622601.
48. Monsanto Co., Pat UK N 1036174.
49. Montecatini Edison s.p.a., Pat. UK N 1130836.
50. Montecatini Edison s.p.a., Pat. France N 95620.
51. Sanhuera E., Hisatsune J.C., Heicklen J. Oxydation of Haloethylenes.-Chemical Reviews, 1976, V.76, N 6, p. 815-821.
52. Heicklen J. the reaction of ozone with perfluoroolefines.- J. Phys. Chem., 1966, V.70, N 2, p. 477-480.
53. Gozzo F., Camaggi G. La reactione tra ozono e tetrafluoroetilene.-Chim.ind.(Milan), 1968, V.50, N 2, p. 197-199.
54. Gozzo F., Bergomi A., Camaggi G. The ozone-induced oxydation of TFE.- Europen Polymer Journal,1970, V.6, N 2, p. 219-232.
55. Toby F.S., Toby S. Kinetic of the reaction of ozone with TFE.- J. Phys. Chem., 1976, V.80, N 21, p. 2313-2316.
56. Agopovich J.W.,Gills C.W. Ozonolysis of trifluoroethylene, 1,1-difluoroethylene and perfluoroethylene. Epoxide and ozonide formation.- J.Am.Chem.Soc.,1980,V.102,N 25,p. 7572-7574.
57. Toby S., Toby F.S. Singlet and triplet emission from difluoromethylene in the reaction of ozone with TFE.-J. Phys. Chem., 1980, v. 84,N 2, p. 206-207.
58. Agopovich J.W.,Gilles C.W. Ozonolysis of 1,1-difluoroethylene, trifluoroethylene and perfluoroethylene.-J.Am.Chem.Soc.,1983,v.105,N 15, p. 5047.
59. Valov P.I., Moiseeva N.I., Sokolov L.F., Sokolov S.V., Blyumberg E.A. Kineticheskie zakonomernosti zhidkofaznogo okisleniya GFP.-Dokl. AN SSSR, 1975, t. 225, N 2, s. 336-341.
60. Kartsev S.V., Valov P.I., Sokolov L.F. Rol' poverhnosti reaktsionnogo sosuda v protsesse zhidkofaznogo okisleniya GFP.- Izv. AN SSSR, 1978, t.10, s. 2268.
61. Kartsev S.V.. Valov P.I., Sokolov L.F., Blyumberg E.A., Sokolov S.V. Zhidkofaznoe okislenie GFP.- Izv. AN SSSR, 1975, t.10, 2230-2235.
62. Valov P.I., Sokolov L.F., Vliyanie skorosti initsiirovaniya i nachal'nyh kontsentratsij reagentov na selektivnost' ehpoksidirovaniya ftorolefinov molekulyarnym kislorodom. -Neftekhimiya, 1982. t.22, N 2, s. 247-253.
63. Pat 1460246 France.
64. Sokolov L.F., Manujlova E.A. i dr. Termicheskoe i geterogenno-kataliticheskoe na serebre okislenie TFEH.-III Vsesoyuznaya konferentsiya po khimii ftororganicheskih soedinenij, Odessa, 1978, Tezisy dokladov, s.57.
65. US Pat. 3775440.
66. Ostrovskij V.E., Kul'kova I.V. i dr. "Kinetika i kataliz", 1964. t.5. s.469-477.
67. Margolis L.Ya. Okislenie uglevodorodov na geterogennyh katalizatorah. M., Khimiya, 1977, s.75-80.
68. Chambers R.D., Mobbs R.G., Ionnye reaktsii perftorolefinov. / V kn. Uspekhi khimii ftora. L., Khimiya. 1970, t.3-4. s.258.
69. J.Catalysts,1974,v. 33, p.392-401.
70. Belen'kij G. G.. German L.S., Knunyants I.L., Izv. AN SSSR, 1968, N 3., s.554-558.
71. Muhametshin F.M. Uspekhi khimii ftororganicheskih gipogalogenitov i rodstvennyh soedinenij., Uspekhi khimii, 1980, t. XI- IX, vyp.7, s.1260-1288.
Fluorine Notes, 1998, 1, 1-2
