Fluorine Notes, 2000, 11, 5-6
Use of perfluoroacylfluorides for the synthesis of perfluoroalkylvinyl ethers. Part II.Perfluorinated ethers of general formula CF3O(CF2O)nCF2CF2OCF=CF2Ershov A.E., Popova L.M.Previous report was devoted to the methods of synthesis of monomers to produce fluoropolymers of general formula CF3O(CF2O)nCF=CF2 with a reduced glass-transition temperature which were produced both by a route of thermal decomposition of salts of appropriate perfluoro-substituted carbonic acids and by thermal interaction of acylfluorides of such acids with oxygen-containing salts of alkaline metals in a medium of solvent and without it. First vinyl ethers (VE) of general formula CF3O(CF2O)nCF2CF2OCF=CF2 (II) were synthesized by P.R.Resnick [2,3]. The author proposes to produce the raw material for the synthesis of vinyl ethers, acylfluorides of CF3O(CF2O)nCF2COF formula (YI) , by electrochemical fluorination of appropriate hydrocarbon analogues or by low-temperature oxidation of tetrafluoroethylene under exposure to an ultraviolet lamp [4]. Both methods are extremely labour-intensive and expensive, the yield of goal low-molecular oligomers of general formula YI is very small due to formation of a great amount of side-products. Tetrafluoroethylene oxidation under ultraviolet exposure requires a long time of contact of reagents with an UV-lamp (30 min) that is the cause of the low productivity ( the yield of a mixture of liquid reaction products is some grams an hour and the yield of the goal compounds of general formula YI in this mixture is only 50-60%). Moreover, a low yield in the every stage of the synthesis of the vinyl ethers (about 20%) obtained by P.R.Resnick should be noted. This study suggests to use side products of pilot process to produce hexafluoropropene oxide (HFPO) by thermal liquid-phase oxidation of hexafluoropropene instead of development of a special method to synthesize acylfluorides VI. These products were collected in considerable quantity in the production of HFPO and did not find any application.
Under process conditions as a result of acylfluoride YII hydrolysis, peroxidates [CF3O(CF2O)nOCOF+CF3O(CF2O)nOCF2COF] and carbonyl fluoride decomposed to CO2 and HF: CF3O(CF2O)nCOF + (n + 2) H2O
and trifluoroacylfluoride and acylfluorides YI were converted to the appropriate acids: CF3O(CF2O)nCF2COF + H2O
We used acids YIII as the raw material to produce VE II in several stages. Stage 1. Synthesis of acylchlorides CF3O(CF2O)nCF2COCl (IX). 2 CF3O(CF2O)nCF2COOH + p-ClCPhCCl
A prevailing method to chlorinate P2O5 was not used in this case because it was difficult to separate the chlorination products from POCl3. The reaction proceeded with hexachloroparaxylol [5] to completion for 5 hours at 200oC even without stirring. Cease of HCl release witnessed about the reaction completion. An absorption band at 1790 cm-1 characteristic for the carbonyl group of perfluorocarbonic acid disappeared in the IR-spectra of the reaction products and a band at 1810cm-1 characteristic for the carbonyl group of acylfluorides appeared. Stage 2. Fluorination of acylfluorides IX. CF3O(CF2O)nCF2COCl (IX) + KF CF3O(CF2O)nCF2COF ( YI) + KCl To replace halogen atoms with fluorine, potassium fluoride in aprotic polar solvents is widely used, the best of the solvents are amides and sulfones, for example tetramethylene sulfon (sulfolan) [6]. Such solvents with high permittivity are able to dissolve noticeable amount of alkaline metals fluorides. High boiling temperatures of the solvents allow to carry out the reaction at high temperature and atmospheric pressure. Acylfluorides of general formula IY were obtained by a reaction of acylchlorides IX with potassium fluoride in sulfolane at 200oC. The fluorination was run to completion for 10 hours. An absorption band of the carbonyl group of fluoroanhydride of perfluorocarbonic acids appeared in the IR-spectrum at 1890 cm-1. Disappearance of the absorption band of the carbonyl group of acylfluorides (at 1810 cm-1) was evidence of the reaction completion. Stage 3. Synthesis of products of addition of hexafluoropropene oxide to acylfluorides VI The interaction of HFPO with acylfluorides VI was carried out by P.R.Resnick according to the following scheme:
Compounds X were produced by P.R.Resnick mainly in a yield of about 50%. In this study,the products of addition X were obtained in a yield of up to 95% as a result of replacement of CsF with KF ( less active catalyst of oligomeric addition of HFPO to acylfluorides) and of a reduction of relative amount of diglyme. We have determined the optimal conditions of addition of one HFPO molecule to acylfluorides first in the synthesis of acylfluorides III [1]. Stage 4. Synthesis of vinyl ethers II. P.R.Resnick in studies [1,2] obtained VE of general formula II by a reaction of acylfluorides X with soda in diglyme. But only most low-molecular vinyl ether with n=1 was produced in a satisfactory yield. The yield of vinyl ether with n>1 either was not shown or was very small ( for example 20% for n=4). In this connection in this study first we used a method to synthesize VE by thermal decomposition of appropriate salts CF3O(CF2O)nCF2CF2OCF(CF3)COONa (XI). But in the pyrolysis of salts XI ( similarly as in the pyrolysis of salts V[1]), the formation of a significant amount of by-products ( acylfluorides X and VI) reducing the yield of VE II to 60% was observed.
The composition and structure of each product of the pyrolysis of salts XI were proved by GLC, IR and 19F NMR spectroscopy. In this investigation we have studied conditions of interaction of acylfluorides X with soda in the presence of a solvent with the purpose to increase the VE yield in comparison with the yields of these products obtained by P.R.Resnick [2,3].
First, the low yields obtained by P.R.Resnick may be obviously explained by incompleteness of the reaction of formation of salt XI when acylfluorides X were added to a dispersion of soda in diglyme. When acylfluorides X are added into the reactor filled with a formed gel of sodium salt XI, the reaction is slowed down and it is necessary to keep the reaction temperature (60oC) by means of external heating to complete the reaction. P.R.Resnick [2,3] obtained the salts mainly at room temperature during 12-16 hours. The second cause of the low VE yield in the mentioned studies is obviously the use of great amount of a solvent (diglyme), i.e. a 7-fold molar excess with respect to acylfluoride X. At a strictly determined ratio of the components and a pyrolysis temperature of 120-140oC in this study , VE II have been obtained in 95% at a conversion of 90-95%. The improvement of the methods to synthesize VE II allowed to increase considerably the yields of these compounds in comparison with the reference data. Thus, as a result of the study of the methods to synthesize perfluoropolyoxaalkylvinyl ethers it has been determined that the best method to synthesize VE of general formula I and II is interaction of 2-(polyoxaalkyl)propionyl fluorides with soda in a solvent while a fair quantity of by products is formed in the synthesis of the same VE by the pyrolysis of the appropriate perfluoro-substituted carbonic acids. Experimental The IR-spectra were recorded on a IKS-29 instrument. The 19F NMR spectra were measured at a 84.67MHz frequency on a Brucker Spectrospin HX-90 instrument using hexafluorobenzene as internal standard. Synthesis of chloroanhydrides of perfluoropolyoxacarbonic acids of general formula CF3O(CF2O)nCF2COCl (IX). 1688g (5.4 mole) of hexachloroparaxylol was added to 2840g(7.2 mole) of a mixture of acids of general formula CF3O(CF2O)nCF2COOH (YIII) with an average molecular mass of 395 and heated at 200oC for 5 hours. 2530g of acylfluorides of general formula IX was obtained in a 85% yield by distillation. The mixture composition was determined by GLC. The individual compounds were isolated by rectification. The boiling temperatures and composition of the mixture are given in the table. Table. Boiling temperatures and composition of the mixture of acylchlorides IX (n=1,2,3,4,5,6)
3,5,7,9-tetraoxaperfluorodecanoylfluoride (VI) (n=3). 450g
of sulfolane and 380g of KF were added to 1800g of 3,5,7,9-teraoxaperfluorodecanoyl
chloride (IX, n=3), the mixture was heated to 200-220oC for 10 hours.
1525g of acylfluoride YI (n=3),b.p.87oC, was obtained by distillation.19F
NMR(CFCl3): 2-Trifluoromethyl-3,6,8,10,12-pentaoxaperfluorotridecanoyl fluoride (X,
n=3) was synthesized under conditions similar to those to obtain compounds IY (m=2)
[1]. B.p.135-136oC. 19F NMR (CFCl3): 2-Trifluoromethyl-3,6,8,10,12-pentaoxaperfluorotridecantanoate of sodium (XI, n=3) and the rest salts of general formula XI were obtained by treatment of appropriate acylfluorides X with 10% aqueous solution of NaOH under conditions similar to those of the synthesis of salts Y (m=2)[1]. The synthesis of 3,6,8,10,12-pentaoxaperfluorotridecene-1 (II, n=3) was carried out by thermal decomposition of salt XI (n=3) at 200oC
under conditions similar to those of the thermal decomposition of salts Y (m=2)[1].
42 g of the equimolar mixture of salt XI (n=3) and NaF was used to obtain 32g
of pyrolyzate containing 64% of 3,6,8,10,12-pentaoxaperfluorotridecene-1 II (n=3),
30% of acylfluoride X (n=3) and 6% of acylfluoride YI (n=3). 20g of VE II (n=3),
b.p.125-125.5oC was isolated by rectification. The yield of VE II
(n=3) attained 60% counting on the initial sodium salt. 19F NMR (CFCl3):
Similarly VE of general formula II were obtained by means of the pyrolysis of rest salts XI at n=1 (b.p.76-78oC, n=2 (b.p.103oC), n=4 (b.p.145oC), n=5 (b.p.157oC) and n=6 (b.p.86oC/23 Torr). Thermal decomposition of salt XI (n=3) in the presence of sodium carbonate. The pyrolysis was carried out at 200oC under the conditions of the previous synthesis. 47.5 g of the equimolar mixture of salt XI (n=3) and NaF was used with addition of dry Na2CO3 to obtain 33.1g of the pyrolyzate which contained mainly VE II (n=3) according to the GLC data. After separation and distillation the yield attained 80% counting on the initial salt. The synthesis of 3,6,8,10,12-pentaoxaperfluorotridecene-1 (II, n=3) by interaction of acylfluoride X (n=3) with soda in the presence of diglyme was carried out under the conditions similar to those of the decomposition of acylfluorides of general formula III[1] from 51.5g of Na2CO3, 28 mL of diglyme and 200g of acylfluoride X. After isolation and distillation the yield of VEII was 91%.VE II for n=1,2,4,5,6, were obtained under similar conditions and approximately in the same yields. References 2. Patent US 3692843. Perfluorovinyl ethers. 3. Patent US 3817960. Polymers of perfluorovinyl ethers. 4. Patent Fr 1499859. Procede de preparation de derives orgoniquesfluores contamant de l'oxygene et produits en resultant. 5. Gubanov V.A., Fedorova G.B.‚ Dolgopol'skij I.M., Gracheva P.E. ZhOrKh. 1973. T.9‚ N 11. c. 2209-2211. 6. Ponomorenko V.A., Krukovskij S.P.‚ Alybina A.Yu. Ftorsoderzhashchie geterotsepnye polimery. M: Nauka. 1973. c. 75. |
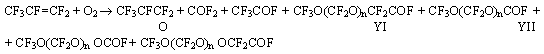
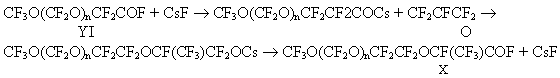
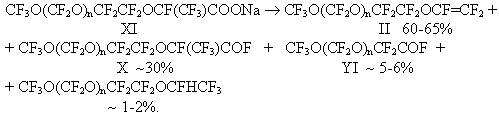
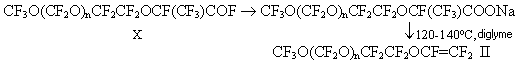
Fluorine Notes, 2000, 11, 5-6
